Shutterstock.com
After reading this article, you should be able to:
- Understand the pathophysiology of cystic fibrosis (CF);
- Explain which organs are affected by CFTR dysfunction;
- Identify the classes of drugs used to manage CFTR dysfunction and understand any considerations for efficacy or adverse effects;
- Understand how health in CF is monitored.
Introduction
Cystic fibrosis (CF) is an inherited, chronic, multisystem disease affecting around 10,900 individuals and 1 in 2,500 live births in the UK. CF is life limiting; the median predicted survival of people born with CF is 53.3 years[1]. It is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. The transport of chloride ions via the CFTR protein into and out of the cells regulates the movement of water necessary to produce free-flowing mucus, which provides a protective coating in organs and tissues, such as the airways and digestive system (see Figure 1)[2,3]. Dehydration and the subsequent viscous mucus on the airway surface results in mucus accumulation, airway obstruction, infection and inflammation, which can lead to loss of lung function, hospitalisation, reduced quality of life and mortality[4–6]. Other manifestations of the disease include pancreatic insufficiency, CF-related diabetes, digestive dysfunction, hepatic failure, chronic sinopulmonary infections, bone disease, growth defects, male infertility and female sub-fertility[7,8].
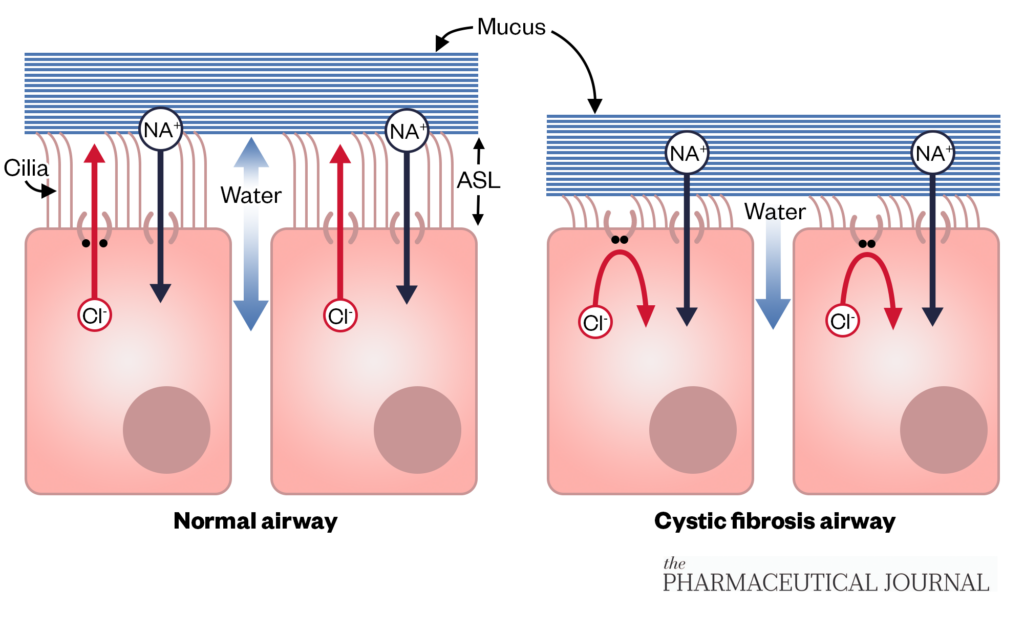
Sodium hyperabsorption and defective chloride secretion drives increased absorption of water by osmosis and reduces the height of the airway surface liquid, leading to impaired muco-ciliary clearance and adherence of mucus to airway epithelial cells
The Pharmaceutical Journal
Treatments to manage the symptoms of CFTR dysfunction are necessary, but regimens are complex, time consuming and lifelong[9,10]. In addition, non-pharmacological treatments, such as chest physiotherapy, exercise, airway clearance and high calorific diets, may be required daily[11].
This article provides an overview of the pharmacological management of respiratory symptoms, highlighting the support that should be given to people with CF to enable them to obtain maximum benefit from their medication regime.
Managing respiratory symptoms
CFTR modulators
Over the past decade, the development of mutation specific therapy known as CFTR modulators (e.g. ivacaftor, ivacaftor + lumacaftor, ivacaftor + tezacaftor and elexacaftor + ivacaftor + tezacaftor) represents a breakthrough in the treatment of CF. This class of drugs corrects the baseline defect in CFTR dysfunction, resulting in improved sodium and chloride transportation across the epithelium[12,13]. CFTR modulators have been shown to improve lung function, nutritional status and stabilisation of the disease in a significant number of people with CF[14,15].
CFTR modulators are pharmacogenomic therapies and their correct use requires genotyping and interpretation of the genomic testing to ensure safety and efficacy. Therefore, before initiation of treatment, a DNA panel of common causing CF-variants must be obtained if not already available. This is vital because CFTR modulators have been licensed and funded for specific variants of the CFTR gene[16,17]. If eligible for treatment, the reasons why treatment is being considered should be discussed with people with CF and their families. Treatment expectations, along with how best to take the medication, what side effects may be experienced and any monitoring required, should also be discussed and backed up with written information. Opportunities for people with CF and their families to discuss any uncertainties or ask any questions should also be encouraged[17].
A detailed medical history, including complementary and alternative therapies, should be taken before initiation of therapy and checked for drug–drug interactions. CFTR modulators are metabolised via the CYP3A system and any inhibitor or inducer of this system can result in increased or reduced plasma levels of the drug, respectively, and may require dosage adjustments[18]. Dose adjustments are also required for patients with severe liver disease[18].
As CFTR modulators have been shown to reduce mucus viscosity, increase ciliary beat frequency and mucociliary transport, some patients experience an increase in sputum production, sometimes referred to as ‘sputum purging’ when treatment is commenced[19]. Transient side effects, such as headache, gastrointestinal symptoms and rashes, usually resolve when doses are reduced or treatment is interrupted[17]. Unexpected side effects, such as insomnia, anxiety and a deterioration in mental health, have been reported with post-marketing surveillance[20,21]. Careful management on an individual patient basis is required if the patient is to remain on treatment[21].
Owing to the complex nature of initiating CFTR modulator therapy treatment, monitoring for efficacy and unwanted adverse effects, ongoing prescribing and provision of the CFTR modulator (via homecare) should only be undertaken by specialist CF teams[16].
More information on the clinical impact of CFTR modulators can be found in ‘Clinical impact of CFTR modulator therapy roll-out’[22].
Respiratory infection
Owing to the characteristic viscous mucus found in the lungs of people with CF, respiratory infection is more common[23]. Impaired mucus transport and mucus adhesion to airway surfaces and these sites on the airway surface become the main site of infection, rather than the airway surface itself (23). Infection induces an excessive and ineffective inflammatory response that contributes to excess mucus cohesion and a chronic cycle of infection and inflammation is initiated, resulting in damage to the structural integrity of the airways and leading to the development of bronchiectasis[24]. Infection of the respiratory tract begins in the first few weeks of life in those with CF[24].
Some of the common pathogens in CF are Staphylococcus aureus, both methicillin sensitive (MSSA) and methicillin resistant (MRSA), Pseudomonas aeruginosa (PA), Burkholderia cepacia complex (BCC) and non-tuberculosis mycobacteria (NTM)[1]. Mycobacterium abscessus complex, a subspecies of NTM, is associated with the most rapid decline in lung function. Of the Gram-negative bacteria, BCC is associated with worse outcomes compared to PA, but there is some interspecies variation[25]. Chronic infection with MRSA is also associated with a more rapid decline in lung function but the impact of MSSA is still debated as there is a challenge distinguishing between true infection and colonisation as carriage rates are high in the healthy population[26,27]. Antibiotics are used to prevent, eradicate or control respiratory infection in CF. The prompt use of effective antibiotics in these situations has been a major reason for the decreased respiratory morbidity and increased longevity seen over recent decades[24].
Long-term oral antibiotics
At the time of diagnosis, infants with CF may be started with a narrow spectrum antibiotic, such as flucloxacillin, up until the age of three years to prevent infection with MSSA, which can be extended to the age of six years, if deemed appropriate[24]. There have been reports that long-term treatment with flucloxacillin could increase the incidence of infection with PA, which is a significantly more problematic pathogen in CF; however, there is currently no evidence to support this[28].
‘CF START: a cystic fibrosis randomised registry trial’ is an ongoing clinical trial investigating the safety and efficacy of long-term antibiotic treatment in infants, including predisposition to PA infection[29]. There are no recommendations for prophylactic flucloxacillin in adults because trial data are not available; however, it is used in adults with CF who frequently grow MSSA and where it is thought that these growths are impacting lung function and/or causing pulmonary exacerbations[30]. This was highlighted by the ‘UK CF registry 2021 annual data report’, published in September 2022, which showed 11% of adults are prescribed long-term prophylactic flucloxacillin[1].
Long-term macrolides are commonly used in CF: azithromycin is prescribed for 56% of people with CF aged 16 years and above[1]. Interestingly, macrolides are used for their anti-inflammatory properties, rather than their antimicrobial action; however, the exact mechanism of this anti-inflammatory action is unknown. Long-term azithromycin has been shown to improve lung function and reduce the frequency of pulmonary exacerbations[31]. There is no consensus on the best dosing regimen, with some centres preferring a low daily dose, while others prefer a higher dose three times a week. Azithromycin is typically given to people with CF infected with PA and most of its evidence of benefit is in this group; however, it has also been proven to be effective regardless of PA infection status[24]. Current recommendations are that azithromycin should be considered for those with CF whose lung function is deteriorating and/or experiencing repeated pulmonary exacerbations[32].
Nebulised and inhaled antibiotics
These have a dual role in CF. First, they are used during attempts to eradicate a pathogen following new isolation in a sputum culture. Second, they are used as chronic suppressive therapy when chronic infection has been established. Nebulised and inhaled antibiotics are mainly used in the treatment of PA, as it is the most common CF pathogen, but they are also used in the treatment of other pathogens, such as BCC and NTM.
There are currently four antibiotics available as a licensed nebulised or inhaled antibiotic for PA infection in CF in the UK: colistimethate (nebulised and inhaled)[33–35]; tobramycin (nebulised and inhaled)[36–39]; aztreonam (nebulised)[40]; and levofloxacin (nebulised)[41].
Early administration of nebulised antibiotics following a newly identified PA isolation in sputum culture or similar has been shown to significantly reduce the risk of chronic infection with PA[42]. Chronic infection is associated with a worse prognosis and a more rapid decline in lung function[29]. Following a new isolation of PA, the clinical team will prescribe eradication therapy. Eradication protocols vary from centre to centre owing to limited data on regimen efficiency[42]. European CF Society best practice guidelines currently recommend up to three months of nebulised colistimethate in combination with oral ciprofloxacin, or one month of nebulised tobramycin for this indication[43]. If the first attempt at eradication fails, marked by a further growth of PA during post treatment sputum surveillance, further endeavours are made owing to the importance of preventing chronic infection. This usually involves using the nebulised antibiotic not used in the first attempt, in combination with oral ciprofloxacin. A two-week course of IV antibiotics can also be used prior to further administration of nebulised antibiotics and oral ciprofloxacin[24].
If all PA eradication attempts fail, then the patient is classed as being chronically infected with this pathogen. The goal of treatment shifts to suppressing the pathogen to improve symptoms associated with chronic respiratory infection, reduce the frequency of pulmonary exacerbations and slow the associated decline in lung function. Nebulised and inhaled antibiotics are used routinely in chronic infection with PA. Figure 2 describes the current treatment pathway[44,45].
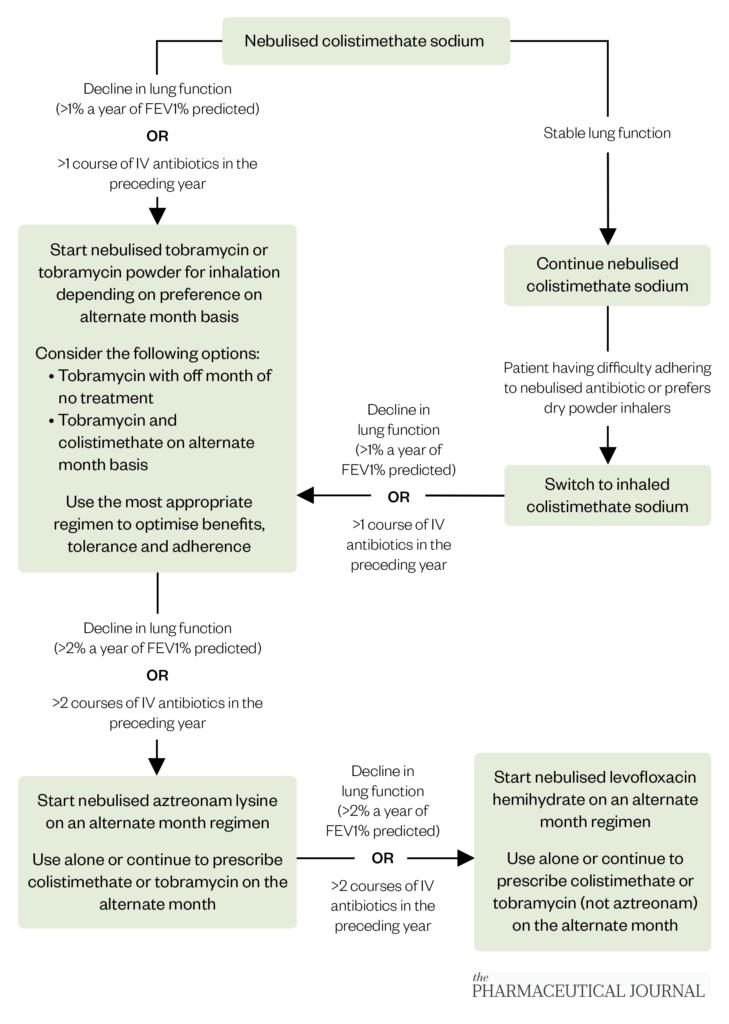
The Pharmaceutical Journal
Only colistimethate is licensed for continuous use, whereas tobramycin, aztreonam and levofloxacin are licensed for use on an alternate month basis[33–41]. This is because the initial clinical trial of tobramycin for chronic PA in CF was done using this alternate month dosing strategy and since the clinical trials for aztreonam and levofloxacin for the same indication followed the same dosing strategy[46–48]. Most centres prescribe a second antibiotic for the off month of the first antibiotic to ensure continuous treatment, but this is dependent on tolerance, adherence and patient preference. Colistimethate and tobramycin come as dry powder inhalers, which are preferred by some people with CF as they have reduced treatment times compared with nebulisation; however, some users of these formulations may develop a dry cough[49]. Nebulised aztreonam and levofloxacin are not currently recommended for eradication purposes as there are no data to support their use.
Nebulised treatments are particularly burdensome in CF, but nebulisers such as the i-neb (Philips) and e-Flow (Pari) drastically reduce treatment times to minimise this burden as much as possible[11].
Pulmonary exacerbations
People with CF can experience episodic flares in their symptoms, known as a pulmonary exacerbation. The exact aetiology of pulmonary exacerbations and the underlying biological mechanisms driving disease are poorly understood[50]. These episodes are typically associated with a combination of the following symptoms:
- Increased cough;
- Increased sputum production and/or viscosity;
- Shortness of breath;
- Reduced lung function on spirometry;
- Decreased energy levels;
- Reduced appetite;
- Weight loss[46].
There is no consensus as to what changes in symptoms are defined as a pulmonary exacerbation and symptoms can range from mild to severe[51]. Therefore, treatment initiation should be based on a detailed history of symptoms and assessing objective measures, such as lung function, weight and blood investigations, as well as a discussion with the person with CF. Milder exacerbations can be treated with oral antibiotics in an attempt to avoid the need for IV treatment and more severe exacerbations will need treatment with IV antibiotics[24]. This could be done either at home or in hospital, depending on the severity and patient preference. Home IV antibiotics are only indicated when a patient, or a relative/carer in the case of children, have been trained and assessed by a CF specialist nurse to administer IV medication in a suitable home environment[24]. Support from a CF specialist nurse should be available for those administering home IV antibiotics and a treatment plan should be put into place to ensure other aspects of care that would be available during hospital admission, such as specialist physiotherapy, are being included as part of the treatment[24,43]. Pre-prepared IV antibiotics, such as those provided by homecare companies, are preferable but reconstitution of IV antibiotics at home by the patient does occur[24].
Empirical treatment is started based on sputum culture history, with treatment aimed at targeting the known chronic pathogens. A respiratory sample, ideally sputum but a cough swab would suffice, should be collected at each clinic visit[24]. This should be obtained before starting antimicrobial treatment and the results can inform future decisions should the patient not adequately respond to the chosen empiric treatment and for future exacerbations[24].
The role of antimicrobial sensitivity testing (AST) is controversial in CF. While it is a cornerstone of infection treatment, the majority of studies have failed to show a relationship between AST and clinical response[52]. Therefore, the sensitivity pattern reported may act as a guide but it should be noted that sensitivity patterns and clinical response may be discordant, and therefore the sensitivity pattern should not relied upon to inform all treatment decisions[24]. Preference may be given to a combination of IV antibiotics to which the patient has responded well when given for previous exacerbations irrespective of sensitivity patterns[24].
For pulmonary exacerbation, treatment courses for both oral and IV antibiotics are typically two weeks. Combination treatment with two IV antibiotics is routine practice, typically a broad-spectrum beta-lactam (e.g. ceftazidime, piperacillin-tazobactam and meropenem) with an aminoglycoside (e.g. tobramycin because it is less nephrotoxic than gentamicin and more efficacious against PA), to take advantage of potential antimicrobial synergy[53,54]. Evidence from a systematic review suggests that combination treatment is less likely to induce resistance compared to monotherapy when treating pathogens like PA[24]. Doses of antibiotics used in CF are typically larger than in the non-CF population, this is especially true for beta-lactams and aminoglycosides because pharmacokinetic differences, such as increased clearance, have been observed in people with CF[55]. Older studies reported that people with CF have a higher volume of distribution compared to healthy controls owing to a relatively high lean body mass, but as weight and nutritional status has improved in CF this is not as relevant[55].
Antimicrobial resistance (AMR) is very much an issue in CF — intrinsic resistance is present in many pathogens observed. For example, PA is resistant to almost all oral antibiotics with only fluoroquinolones and chloramphenicol noted to have activity against PA, while ciprofloxacin is the fluoroquinolone of choice in CF[24,56]. Pathogens such as BCC, NTM, Achromobacter sp. and Stenotrophomas maltophilia can be intrinsically multi-drug resistant. Acquired resistance can occur through either mutation or horizontal gene transfer and fluoroquinolone resistance in PA is an example of acquired AMR[57].
Adaptive AMR mechanisms have also been observed, such as the formation of biofilms by PA[58]. Biofilms are aggregates of micro-organisms that adhere to each other on a surface and are embedded within a self-produced extracellular matrix[58]. Bacteria within biofilms are inherently more resistant to antimicrobials as the extracellular matrix slows the penetration of antibiotics, the altered micro-environment within the biofilm slows bacterial growth which reduce antibiotic uptake and multidrug resistant persister cells can be formed[58]. Combined AMR mechanisms mean that chronic infections in CF can become harder to treat with conventional treatments over time because the infecting pathogen becomes increasingly resistant to treatment with previously used antimicrobials. When this occurs, new generation beta-lactam/beta-lactamase inhibitor combinations, such as ceftolozane/tazobactam and ceftazidime/avibactam, are considered for use[59].
Despite the lung inflammation that characterises CF, corticosteroids are not routinely used to treat pulmonary exacerbations. This owes to limited evidence of benefit, meaning current consensus documents advise against routine use for pulmonary exacerbations until more data are available[60]. Furthermore, two studies published in 2021 and 2023, respectively, completed in paediatric populations have shown no benefit of corticosteroid use[61,62]. However, it is noted that some people with CF may benefit from a short course of corticosteroids when acute severe chest tightness or wheezing is present during an exacerbation[60,63].
Chest clearance
People with CF should perform airway clearance techniques at least daily to help keep their lungs clear of mucus. There are various airway clearance techniques, which differ in assistance or equipment needs and cost. Physiotherapy techniques include:
- Active cycle of breathing techniques — a combination of breathing control, thoracic expansion exercises and forced expiration exercises;
- Autogenic drainage — uses controlled breathing at different lung volumes to loosen, mobilise and move secretions in three stages towards the larger central airways;
- Postural drainage — gravity assisted positioning helps drainage of secretions from some areas of the lungs;
- Positive expiratory pressure (PEP) — a machine produces positive pressure in the airways to open them and help move secretions higher up the respiratory tract;
- Oscillating PEP – similar to PEP but adds resistance to expiratory flow causing vibrations to the airways to loosen sputum[64].
Results from a 2019 Cochrane review showed that there is little evidence to support the use of one airway clearance technique over another; therefore, people with CF should choose the airway clearance technique that best meets their needs, after considering many factors including comfort, convenience, flexibility, practicality and cost[65]. This decision should be guided by a CF specialist physiotherapist as they are best placed to help pinpoint which techniques are best for each individual and assess the effectiveness of the chosen modality.
Mucolytics are a useful adjuvant to airway clearance to aid sputum expectoration. They reduce the viscosity of CF sputum, making it easier to clear in combination with the chosen airway clearance technique. All mucolytics in CF have been shown to improve lung function and reduce the number of pulmonary exacerbations; however, NICE recommends dornase alfa as first-line treatment[32,66–68].
Dornase alfa is a naturally occurring enzyme that reduces sputum viscosity by cleaving extracellular DNA in sputum[69]. It is a nebulised treatment, from which most benefit is obtained from once-a-day dosing, although this can be increased to twice a day if needed. The increased frequency of administration has been associated with a reduced risk of pulmonary exacerbation, requiring parenteral antibiotics[70].
Hypertonic saline, an osmotic mucolytic, is the second-line mucolytic of choice in CF[32]. It can be used either alone or in combination with dornase alfa and is also nebulised[32]. Varying strengths are available ranging from 3–7% and CF teams will prescribe the highest tolerated strength for the individual. Mannitol dry powder for inhalation, also an osmotic mucolytic, can be used in patients who cannot tolerate or are unresponsive to dornase alfa and cannot tolerate hypertonic saline[32].
Since the widespread use of highly effective CFTR modulators, sputum production has significantly dropped, even in those with advanced lung disease[71]. This has led to questions as to whether mucolytics still offer the same benefit. Ongoing trials are investigating whether mucolytics can be safely stopped in patients taking CFTR modulators. In 2022, the ‘SIMPLIFY’ trial reported no meaningful differences in lung function in those who stopped treatment after six weeks compared to those who continued; however, the long-term impact of stopping such treatments is still unknown[72]. ‘CF STORM’ is assessing the impact of stopping mucolytics in this cohort over 12 months[73].
Monitoring
Pulmonary function of people with CF should be monitored at each clinic visit. Current recommendations are that the frequency of clinic reviews should be based a person’s clinical condition, but children and young people should be reviewed at least every eight weeks and adults every three months[32]. It should be noted that these recommendations were made before the widespread use of CFTR modulators in the UK. At each review the following should be completed[32,74]:
- Lung function testing, using spirometry for adults and children who can complete the test;
- Measurement of oxygen saturation;
- Collection of respiratory secretion sample for microbiological testing;
- A clinical assessment including a review of clinical history and medicines adherence (reviewed based on patient’s self-reports and/or assessing medication possession ratio).
Sputum is the preferred sample for microbiological screening[32]. They can be obtained either spontaneously or via the induced sputum method which involves nebulising hypertonic saline to aid sputum expectoration[75]. Other respiratory secretion samples that can be used include cough swabs, nasal pharyngeal aspirate (NPA) and bronchoalveolar lavage (BAL)[32,74]. Sputum is the preferred as it has been shown to be more effective at detecting respiratory pathogens than cough swabs and NPA, and it is significantly less invasive to collect than BAL which requires bronchoscopy[76,77].
Summary
Pharmacological treatment regimens to manage the effects of CFTR dysfunction on respiratory system are complex and require careful monitoring for efficacy and unwanted effects. The regimes are time consuming, lifelong and have a large impact on patient’s quality of life[10]. However, pharmacy professionals are well placed to provide support and advice to enable maximum benefit to be obtained in a safe and effective manner.
- 1UK Cystic Fibrosis Registry 2021 Annual Data Report. Cystic Fibrosis Trust. 2022.https://www.cysticfibrosis.org.uk/sites/default/files/2022-10/CFT_2021-Annual-Data-Report-WEB.pdf (accessed May 2023).
- 2Riordan JR. CFTR Function and Prospects for Therapy. Annu. Rev. Biochem. 2008;77:701–26. doi:10.1146/annurev.biochem.75.103004.142532
- 3Stoltz DA, Meyerholz DK, Welsh MJ. Origins of Cystic Fibrosis Lung Disease. N Engl J Med. 2015;372:351–62. doi:10.1056/nejmra1300109
- 4Goss CH, Burns JL. Exacerbations in cystic fibrosis {middle dot} 1: Epidemiology and pathogenesis. Thorax. 2007;62:360–7. doi:10.1136/thx.2006.060889
- 5Britto MT, Kotagal UR, Hornung RW, et al. Impact of Recent Pulmonary Exacerbations on Quality of Life in Patients With Cystic Fibrosis. Chest. 2002;121:64–72. doi:10.1378/chest.121.1.64
- 6Liou TG, Adler FR, FitzSimmons SC, et al. Predictive 5-Year Survivorship Model of Cystic Fibrosis. American Journal of Epidemiology. 2001;153:345–52. doi:10.1093/aje/153.4.345
- 7Hughan KS, Daley T, Rayas MS, et al. Female reproductive health in cystic fibrosis. Journal of Cystic Fibrosis. 2019;18:S95–104. doi:10.1016/j.jcf.2019.08.024
- 8White H, Chadwick H, Shaw N, et al. Evaluation of an RCT web-based intervention for adherence in cystic fibrosis. Indianapolis: 2017.
- 9Sawicki GS, Tiddens H. Managing treatment complexity in cystic fibrosis: Challenges and Opportunities. Pediatr. Pulmonol. 2012;47:523–33. doi:10.1002/ppul.22546
- 10Davies G, Rowbotham NJ, Smith S, et al. Characterising burden of treatment in cystic fibrosis to identify priority areas for clinical trials. Journal of Cystic Fibrosis. 2020;19:499–502. doi:10.1016/j.jcf.2019.10.025
- 11Sawicki GS, Sellers DE, Robinson WM. High treatment burden in adults with cystic fibrosis: Challenges to disease self-management. Journal of Cystic Fibrosis. 2009;8:91–6. doi:10.1016/j.jcf.2008.09.007
- 12Ramsey BW, Davies J, McElvaney NG, et al. A CFTR Potentiator in Patients with Cystic Fibrosis and theG551DMutation. N Engl J Med. 2011;365:1663–72. doi:10.1056/nejmoa1105185
- 13Bell SC, De Boeck K, Amaral MD. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacology & Therapeutics. 2015;145:19–34. doi:10.1016/j.pharmthera.2014.06.005
- 14Volkova N, Moy K, Evans J, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. Journal of Cystic Fibrosis. 2020;19:68–79. doi:10.1016/j.jcf.2019.05.015
- 15Duckers J, Lesher B, Thorat T, et al. Real-World Outcomes of Ivacaftor Treatment in People with Cystic Fibrosis: A Systematic Review. JCM. 2021;10:1527. doi:10.3390/jcm10071527
- 16Clinical Commissioning Urgent Policy Statement Cystic Fibrosis Modulator Therapies Access Agreement for licensed mutations [200810P]. NHS England. 2020.https://www.england.nhs.uk/wp-content/uploads/2020/08/Policy-statement-CFTR-Modulator-Therapies-Licensed-mutations.pdf (accessed May 2023).
- 17Southern KW, Castellani C, Lammertyn E, et al. Standards of care for CFTR variant-specific therapy (including modulators) for people with cystic fibrosis. Journal of Cystic Fibrosis. 2023;22:17–30. doi:10.1016/j.jcf.2022.10.002
- 18Kaftrio – summary of product characteristics. European Medicine Agency. https://www.ema.europa.eu/en/medicines/human/EPAR/kaftrio (accessed May 2023).
- 19Birket SE, Chu KK, Houser GH, et al. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2016;310:L928–39. doi:10.1152/ajplung.00395.2015
- 20Heo S, Young DC, Safirstein J, et al. Mental status changes during elexacaftor/tezacaftor / ivacaftor therapy. Journal of Cystic Fibrosis. 2022;21:339–43. doi:10.1016/j.jcf.2021.10.002
- 21Spoletini G, Gillgrass L, Pollard K, et al. Dose adjustments of Elexacaftor/Tezacaftor/Ivacaftor in response to mental health side effects in adults with cystic fibrosis. Journal of Cystic Fibrosis. 2022;21:1061–5. doi:10.1016/j.jcf.2022.05.001
- 22Clinical impact of CFTR modulator therapy roll-out. Pharmaceutical Journal. 2022. doi:10.1211/pj.2022.1.158712
- 23Turcios NL. Cystic Fibrosis Lung Disease: An Overview. Respir Care. 2019;65:233–51. doi:10.4187/respcare.06697
- 24Antibiotic treatment for cystic fibrosis. Cystic Fibrosis Trust. 2009.https://www.cysticfibrosis.org.uk/the-work-we-do/resources-for-cf-professionals/consensus-documents (accessed May 2023).
- 25Langton Hewer SC, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database of Systematic Reviews. 2017;2020. doi:10.1002/14651858.cd004197.pub5
- 26Dasenbrook EC. Association Between Respiratory Tract Methicillin-Resistant S<emph type=”ital”>taphylococcus aureus</emph> and Survival in Cystic Fibrosis. JAMA. 2010;303:2386. doi:10.1001/jama.2010.791
- 27Hurley MN. Staphylococcus aureus in cystic fibrosis: problem bug or an innocent bystander? Breathe. 2018;14:87–90. doi:10.1183/20734735.014718
- 28Rosenfeld M, Rayner O, Smyth AR. Prophylactic anti-staphylococcal antibiotics for cystic fibrosis. Cochrane Database of Systematic Reviews. 2020;2020. doi:10.1002/14651858.cd001912.pub5
- 29CF START – A Cystic Fibrosis Randomised Registry Trial . CF START. https://www.cfstart.org.uk (accessed May 2023).
- 30Ahmed MI, Mukherjee S. Treatment for chronic methicillin-sensitive Staphylococcus aureus pulmonary infection in people with cystic fibrosis. Cochrane Database of Systematic Reviews. 2018;2018. doi:10.1002/14651858.cd011581.pub3
- 31Southern KW, Barker PM, Solis-Moya A, et al. Macrolide antibiotics for cystic fibrosis. Cochrane Database of Systematic Reviews. 2012. doi:10.1002/14651858.cd002203.pub4
- 32Cystic fibrosis: diagnosis and management. National Institute for Health and Care Excellence. 2017.https://www.nice.org.uk/guidance/ng78/resources/cystic-fibrosis-diagnosis-and-management-pdf-1837640946373 (accessed May 2023).
- 33Colomycin 2 million International Units (IU) Powder for solution for injection, infusion or inhalation – Summary of Product Characteristics . Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/9664/smpc (accessed May 2023).
- 34Promixin 1 million International Units (IU) Powder for Nebuliser Solution – Summary of Product Characteristics . Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/4 (accessed May 2023).
- 35Colobreathe 1,662,500 IU inhalation powder, Hard Capsules – Summary of Product Characteristics . Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/3063 (accessed May 2023).
- 36Tobi Podhaler 28 mg inhalation powder, hard capsules – Summary of Product Characteristics . Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/4757 (accessed May 2023).
- 37Bramitob 300 mg/4ml Nebuliser Solution – Summary of Product Characteristics . Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/6451 (accessed May 2023).
- 38Vantobra 170mg nebuliser solution – Summary of Product Characteristics. Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/7362/smpc (accessed May 2023).
- 39Tobi 300 mg/5 ml Nebuliser Solution – Summary of Product Characteristics . Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/262/smpc (accessed May 2023).
- 40Cayston 75 mg powder and solvent for nebuliser solution – Summary of Product Characteristics . Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/4456 (accessed May 2023).
- 41Quinsair 240 mg nebuliser solution – Summary of Product Characteristics . Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/7202 (accessed May 2023).
- 42Qvist T, Taylor-Robinson D, Waldmann E, et al. Comparing the harmful effects of nontuberculous mycobacteria and Gram negative bacteria on lung function in patients with cystic fibrosis. Journal of Cystic Fibrosis. 2016;15:380–5. doi:10.1016/j.jcf.2015.09.007
- 43Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. Journal of Cystic Fibrosis. 2018;17:153–78. doi:10.1016/j.jcf.2018.02.006
- 44Clinical Commissioning Policy: Inhaled Therapy for Adults and Children with Cystic Fibrosis. NHS England . 2014.https://www.england.nhs.uk/commissioning/wp-content/uploads/sites/12/2015/01/a01-policy-inhld-thrpy-cf.pdf (accessed May 2023).
- 45Clinical Commissioning Policy: Levofloxacin nebuliser solution for chronic Pseudomonas lung infection in cystic fibrosis (adults). . NHS England. 2018.https://www.england.nhs.uk/wp-content/uploads/2018/08/Levofloxacin-nebuliser-solution-for-chronic-Pseudomonas-lung-infection-in-cystic-fibrosis-adults.pdf (accessed May 2023).
- 46Ramsey BW, Pepe MS, Quan JM, et al. Intermittent Administration of Inhaled Tobramycin in Patients with Cystic Fibrosis. N Engl J Med. 1999;340:23–30. doi:10.1056/nejm199901073400104
- 47McCoy KS, Quittner AL, Oermann CM, et al. Inhaled Aztreonam Lysine for Chronic Airway Pseudomonas aeruginosa in Cystic Fibrosis. Am J Respir Crit Care Med. 2008;178:921–8. doi:10.1164/rccm.200712-1804oc
- 48Stuart Elborn J, Geller DE, Conrad D, et al. A phase 3, open-label, randomized trial to evaluate the safety and efficacy of levofloxacin inhalation solution (APT-1026) versus tobramycin inhalation solution in stable cystic fibrosis patients. Journal of Cystic Fibrosis. 2015;14:507–14. doi:10.1016/j.jcf.2014.12.013
- 49Uttley L, Harnan S, Cantrell A, et al. Systematic review of the dry powder inhalers colistimethate sodium and tobramycin in cystic fibrosis. European Respiratory Review. 2013;22:476–86. doi:10.1183/09059180.00001513
- 50Stanford GE, Dave K, Simmonds NJ. Pulmonary Exacerbations in Adults With Cystic Fibrosis. Chest. 2021;159:93–102. doi:10.1016/j.chest.2020.09.084
- 51Goss CH. Acute Pulmonary Exacerbations in Cystic Fibrosis. Semin Respir Crit Care Med. 2019;40:792–803. doi:10.1055/s-0039-1697975
- 52Somayaji R, Parkins MD, Shah A, et al. Antimicrobial susceptibility testing (AST) and associated clinical outcomes in individuals with cystic fibrosis: A systematic review. Journal of Cystic Fibrosis. 2019;18:236–43. doi:10.1016/j.jcf.2019.01.008
- 53Klastersky J, Daneau D, de Maertelaer V. Comparative study of tobramycin and gentamicin with special reference to anti-Pseudomonas activity. Clinical Pharmacology & Therapeutics. 1973;14:104–11. doi:10.1002/cpt1973141104
- 54Ratjen F, Brockhaus F, Angyalosi G. Aminoglycoside therapy against Pseudomonas aeruginosa in cystic fibrosis: A review. Journal of Cystic Fibrosis. 2009;8:361–9. doi:10.1016/j.jcf.2009.08.004
- 55Akkerman-Nijland AM, Akkerman OW, Grasmeijer F, et al. The pharmacokinetics of antibiotics in cystic fibrosis. Expert Opinion on Drug Metabolism & Toxicology. 2020;17:53–68. doi:10.1080/17425255.2021.1836157
- 56Oliphant C, Green G. Quinolones: a comprehensive review. Am Fam Physician 2002;65:455–64.https://www.ncbi.nlm.nih.gov/pubmed/11858629
- 57Kidd TJ, Canton R, Ekkelenkamp M, et al. Defining antimicrobial resistance in cystic fibrosis. Journal of Cystic Fibrosis. 2018;17:696–704. doi:10.1016/j.jcf.2018.08.014
- 58Pang Z, Raudonis R, Glick BR, et al. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnology Advances. 2019;37:177–92. doi:10.1016/j.biotechadv.2018.11.013
- 59Haines RR, Putsathit P, Hammer KA, et al. Activity of newest generation β-lactam/β-lactamase inhibitor combination therapies against multidrug resistant Pseudomonas aeruginosa. Sci Rep. 2022;12. doi:10.1038/s41598-022-21101-x
- 60Flume PA, Mogayzel PJ Jr, Robinson KA, et al. Cystic Fibrosis Pulmonary Guidelines. Am J Respir Crit Care Med. 2009;180:802–8. doi:10.1164/rccm.200812-1845pp
- 61Muirhead CA, Lanocha N, Markwardt S, et al. Evaluation of rescue oral glucocorticoid therapy during inpatient cystic fibrosis exacerbations. Pediatric Pulmonology. 2020;56:891–900. doi:10.1002/ppul.25204
- 62Davis CS, Faino AV, Onchiri F, et al. Systemic Corticosteroids in the Management of Pediatric Cystic Fibrosis Pulmonary Exacerbations. Annals ATS. 2023;20:75–82. doi:10.1513/annalsats.202203-201oc
- 63Steroid treatment in cystic fibrosis Factsheet. Cystic Fibrosis Trust. 2015.https://www.cysticfibrosis.org.uk/sites/default/files/2020-11/Factsheet%20Steroid%20Treatment%202016.pdf (accessed May 2023).
- 64Aragon Cuevas O. Cystic fibrosis pathophysiology and management. Clin Pharm 2011;:239–44.
- 65Wilson LM, Morrison L, Robinson KA. Airway clearance techniques for cystic fibrosis: an overview of Cochrane systematic reviews. Cochrane Database of Systematic Reviews. 2019;2019. doi:10.1002/14651858.cd011231.pub2
- 66Yang C, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database of Systematic Reviews. 2021;2021. doi:10.1002/14651858.cd001127.pub5
- 67Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database of Systematic Reviews. 2018;2018. doi:10.1002/14651858.cd001506.pub4
- 68Nevitt SJ, Thornton J, Murray CS, et al. Inhaled mannitol for cystic fibrosis. Cochrane Database of Systematic Reviews. 2020;2020. doi:10.1002/14651858.cd008649.pub4
- 69Pulmozyme 2500 U/ 2.5ml, nebuliser solution – Summary of Product Characteristics . Electronic Medicines Compendiums. https://www.medicines.org.uk/emc/product/1112/smpc#gref (accessed May 2023).
- 70Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. N Engl J Med. 1994;331:637–42. doi:10.1056/nejm199409083311003
- 71Kos R, Neerincx AH, Fenn DW, et al. Real‐life efficacy and safety of elexacaftor/tezacaftor/ivacaftor on severe cystic fibrosis lung disease patients. Pharmacology Res & Perspec. 2022;10. doi:10.1002/prp2.1015
- 72Mayer-Hamblett N, Ratjen F, Russell R, et al. Discontinuation versus continuation of hypertonic saline or dornase alfa in modulator treated people with cystic fibrosis (SIMPLIFY): results from two parallel, multicentre, open-label, randomised, controlled, non-inferiority trials. The Lancet Respiratory Medicine. 2023;11:329–40. doi:10.1016/s2213-2600(22)00434-9
- 73CF STORM. CF STORM. https://www.cfstorm.org.uk/ (accessed May 2023).
- 74Standards for the clinical care of children and adults with cystic fibrosis in the UK. Cystic Fibrosis Trust. 2011.https://www.cysticfibrosis.org.uk/the-work-we-do/resources-for-cf-professionals/consensus-documents (accessed May 2023).
- 75Chanez P, Holz O, et al. Sputum induction. European Respiratory Journal. 2002;20:3S – 8s. doi:10.1183/09031936.02.00000302
- 76Schultz A, Caudri D. Cough swabs less useful but induced sputum very useful in symptomatic older children with cystic fibrosis. The Lancet Respiratory Medicine. 2018;6:410–1. doi:10.1016/s2213-2600(18)30183-8
- 77Eyns H, Piérard D, De Wachter E, et al. Respiratory Bacterial Culture Sampling in Expectorating and Non-expectorating Patients With Cystic Fibrosis. Front. Pediatr. 2018;6. doi:10.3389/fped.2018.00403