ANTONIA REEVE / SCIENCE PHOTO LIBRARY
After reading this article, you should be able to:
- Understand the impact of CF on different organ systems;
- Identify the available pharmacological treatment options to manage this dysfunction;
- Know the monitoring requirements for each condition.
Introduction
Cystic fibrosis (CF) is often thought of as a respiratory disease; however, CF transmembrane conductance regulator (CFTR) dysfunction affects multiple organs systems throughout the body, including the pancreas, liver, gastrointestinal tract and reproductive system[1,2]. More information on the aetiology of the disease and its impact on the respiratory system can be found in the previous article in this series: ‘Cystic fibrosis: management and monitoring of respiratory manifestations‘.
To ensure that holistic care is delivered to the person with CF, it is important that pharmacy professionals understand the effects of CFTR dysfunction on these multiple organ systems. They should also be able to describe the treatment options available to manage the resulting symptoms and any monitoring requirements to ensure treatment effectiveness.
This article outlines the effects of CFTR dysfunction on the pancreas, liver and gastrointestinal tract. It also discusses the pharmacological management of these symptoms and describes the monitoring required for treatment optimisation.
Pancreatic insufficiency
Pancreatic insufficiency (PI) occurs in around 85% of people with CF. Signs and symptoms include[3]:
- Failure to thrive in infants;
- Unexplained weight loss in adults;
- Gas;
- Bloating;
- Dyspepsia;
- Steatorrhea (foul-smelling oily stools that are difficult to flush)[4].
The introduction of newborn screening for CF in the UK has reduced the likelihood of failure to thrive and malabsorption being one of the first signs of the disease in infants[4].
The pancreas is made up of exocrine tissue comprising acinar cells, which secrete pancreatic enzymes, and ductal cells that secrete a bicarbonate rich solution[5]. In people with CF, pancreatic secretions are altered, leading to obstruction of small pancreatic ducts and acini. This causes inflammation, fibrosis and fatty infiltration of the pancreas, which can occur as early as 17 weeks in utero[6]. The most common test used to screen for pancreatic insufficiency is the quantification of faecal elastase (Fel-1) in the stool. Fel-1 is a proteolytic enzyme secreted by the pancreas and low levels (<200micrograms/g stool) are indicative of PI[7]. It is recommended that pancreatic status is confirmed by clinical assessment and Fel-1 measurement at diagnosis. It is also recommended that people with CF who are pancreatic sufficient are assessed annually, with the test repeated if growth is inadequate[8]. The annual assessment should also include the measurement of fat soluble vitamins[9].
Management
Untreated PI will lead to malabsorption and maldigestion of intestinal fat leading to poor nutritional status and a deficiency of fat-soluble vitamins[9]. As a result, replacement in the form of pancreatic enzyme replacement therapy (PERT), fat soluble vitamins and the optimisation of nutrition are the recommended treatment. This should be managed by a dietitian specialised in CF care; however, the pharmacy team also has a role to play in ensuring treatment is optimised for each individual person. This includes ensuring patients are prescribed the appropriate strength of preparation and have sufficient knowledge about each medication.
Pancreatic enzyme replacement therapy
PERT contains protease, lipase and amylase to assist with the digestion of protein, fat and starch and is available in a range of strengths and preparations, allowing the appropriate dose to be given to people with CF of all ages[10]. It is recommended that enteric-coated enzymes are used to avoid destruction by gastric acid[4].
Dosing of PERT is based on body weight, fat content of meals, or pancreatic lipase output[4]. This is because the majority of lipase is produced by the pancreas, while amylase and protease are secreted at other sites in the gastrointestinal tract[9]. Data from cohort studies and audits of clinical practice suggest doses of 500–4,000 units of lipase per gram of dietary fat or 500–2,500 lipase units/kg/meal and 250–1,250 lipase units/kg/snack are recommended in adults and children with CF for optimal absorption of fat and other nutrients[11–14]. Initial dosing should be started at the lower end of the dosage range and titrated based on gastrointestinal symptoms, growth and weight gain[9,15]. PERT should be taken with all food and drinks that contain fat and should be determined on an individual basis[9]. Dosing should be reviewed by a specialist CF dietitian who can accurately assess food and fat intake and advise on dose adjustments as necessary[9].
See Box for practical advice that can be given to people with CF prescribed PERT.
Box: Practical advice for people with cystic fibrosis receiving pancreatic enzyme replacement therapy
- Enzymes should be taken with all food and drinks that contain fat;
- Enzymes are not needed with foods or drinks that do not contain fat, such as cordials, fizzy drinks, fruit juice, fruit or boiled/jelly sweets;
- Enzyme doses should be titrated according to dietary fat intake. Consumption of certain low-fat foods containing high amounts of protein, such as protein shakes, low-fat yogurts and lean meat, may require PERT;
- Enzyme doses should be individualised and people with CF should be taught to adjust doses depending upon the quantity and content of food consumed and on their gastrointestinal symptoms and nutritional status;
- Enzyme requirements will vary considerably between individuals;
- People with CF who have a poor response to PERT or high dosage requirements may benefit from changing the timing of PERT administration;
- Enzyme preparations should be changed to suit needs for example progress to standard strength preparations when a young child is able to swallow capsules; move on to higher strength preparations when enzyme capsule number is inconvenient. When changing to higher strength preparations, it is very important to maintain a similar lipase dose;
- Enzyme doses should not be increased if adherence is poor;
- Adequate hydration is important when taking enzymes.
Other aims of PERT are to achieve normal bowel habits and stool characteristics, as well as to ensure fat soluble vitamin and essential fatty acid status. Fat-soluble vitamins should be replaced with dose adjustments informed by serum levels[16]. See Table 2 for recommended starting doses. It should be noted that there are multicomponent preparations available that contain the fat-soluble vitamins A, D, E and K (Paravit-CF) plus several other vitamins and trace elements[17]. These multi-ingredient preparations can decrease the tablet burden in people with CF; however, as with all fixed dose preparations, they may not be suitable for all people with CF.
CF diabetes
CF diabetes (CFD) is a distinct type of diabetes, separate from type 1 and type 2 diabetes mellitus but sharing certain clinical features with both. Characteristics unique to CFD include the partial loss or dysfunction of pancreatic islets leading to deficiency but not absence of insulin secretion and fluctuating levels of insulin resistance owing to chronic inflammation, which can flare during exacerbations[18]. Onset is often insidious with many individuals being asymptomatic at diagnosis. A drop in weight and lung function can be a sign of undiagnosed CFD. Ketoacidosis is a rare occurrence in CFD[19].
CFD is a well-recognised and common complication of CF, occurring in around 19% of adolescents and 40–50% of adults with CF[18]. It is well documented that CFD drives excessive pulmonary morbidity and mortality in people with CF and those with CFD are at significantly increased risk for earlier mortality than people with CF without diabetes owing to increased rate of pulmonary decline[20]. The majority of people with CFD are pancreatic insufficient but it can occur in those who are pancreatic sufficient, particularly after episodes of pancreatitis[19]. Pancreatitis is rare in CF with an overall prevalence of 1.2%; however, it is more common in those who are pancreatic sufficient with a rate of occurrence of 10.3% compared to 0.5% in people with CF who are pancreatic insufficient[21].
Screening
Current guidance is that screening for CFD should be done annually from the age of 10 years. Previously this was done exclusively using the oral glucose tolerance test (OGTT), where 75g of oral glucose is administered during a fasted state with blood glucose levels taken at 0 and 120 minutes to assess the impact of the glucose on blood levels[19]. A two-hour blood glucose ≥11.1mmol/L is indicative of diabetes[18].
Recent advancements in blood glucose monitoring have led to some centres moving away from the OGTT as the preferred method of screening in favour of continuous glucose monitoring (CGM), which does not require fasting and can monitor glucose levels all day while the individual is at home eating their regular diet and completing their regular activities. However, there is currently no published guidance as to what constitutes a diagnosis of CFD following an abnormal CGM report[19]. Centres using CGM as a screening tool currently look at measures including time spent with a blood glucose >7.8mmol/L and number of excursions >11.0mmol/L[19].
Management
Insulin is the treatment of choice in CFD[18,19]. Regimens utilised include:
- Once daily long-acting insulin monotherapy;
- Pre-meal short acting insulin monotherapy;
- Multiple dose regimens (can be either using biphasic insulins or a basal bolus)[22].
Continuous subcutaneous insulin infusions (insulin pumps) have also been utilised in CFD. There are no trials available comparing the effectiveness of different insulin regimens in CFD[22]. Therefore, the regimen used should be tailored to the timing and severity of glucose excursions in each individual[19,22]. In recent years, there has been growing interest in the use of oral antidiabetic agents, owing to their treatment burden advantage over insulin; repaglinide is the most studied.
In 2020, results from a Cochrane review highlighted three trials comparing repaglinide to insulin and concluded that neither therapy offered an advantage over the other in controlling blood glucose[23]. Other oral agents such as metformin, sulphonylureas and DPP-4 inhibitors have been used in the treatment of CFD and as interest continues to grow on the use of oral agents, clarity on their effectiveness and place in treatment will develop[19,22].
There are no defined targets for glycaemic control in CFD, but good glycaemic control has been shown to reduce pulmonary exacerbations and mortality[19]. Therefore, the aim is to keep blood glucose and HbA1c within normal range while keeping the risk of hypoglycaemia low[19]. Serial blood HbA1c monitoring is done routinely to assess glycaemic control, usually at a minimum of three monthly intervals, but it should be noted that two thirds of people with confirmed CFD have normal HbA1c values[24]. People with CFD are at similar risk to microvascular complications as those with type 1 or type 2 diabetes mellitus if glycaemic control is poor. Therefore, foot and retinopathy screening should take place annually for those with CFD in line with diabetes guidance[19]. Macrovascular complications are rarely reported in those with CFD and, while there are a few case reports of cardiovascular disease in people with CFD, none have been associated with death[18]. However, screening for macrovascular complications should also be done annually owing to the potential for prevalence to increase as CF survival continues to improve[19].
CF liver disease
A leading cause of mortality and morbidity in both adults and children, CF associated liver disease (CFLD) is an organ manifestations of CFTR dysfunction[25].
CFTR is expressed in the apical membrane of cholangiocytes but not hepatocytes, in the liver[26]. CFTR dysfunction leads to a more acidic and viscous bile causing biliary obstruction, an increased concentration of toxic bile acids leading to fibrosis and cirrhosis[25,27].
Multilobular cirrhosis develops in the first decade of life in around 5–10% of patients[27,28]. This can progress to portal hypertension with variceal bleeding in a proportion of patients, requiring intervention and ultimately liver transplantation[29]. UK registry data collected in 2021 showed that the overall incidence of CFLD was 15.3%, with an incidence of 9.3% patients <16 years and 18.9% in patients ≥16 years[30].
The pathogenesis of CLFD is multifactorial and has primary, secondary and tertiary causes:
- Primary causes: genetic factors affecting bile acid homeostasis;
- Secondary causes: CF-related diabetes, pancreatic insufficiency, intestinal inflammation, changes in gut microbiome;
- Tertiary causes: high fat diet, hepatotoxic medication, such as antibiotics[25].
Management
Current best practice guidelines for the diagnosis and management of CFLD recommend starting treatment with ursodeoxycholic acid (UDCA), a naturally occurring hydrophilic bile acid, as soon as the diagnosis of CFLD is made, with the aim of delaying the progression of the disease[16]. A higher than usual dose of UDCA at 20mg/kg/day in divided doses is recommended because of the poor absorption in people with CF owing to a lower small intestinal pH[31,32]. Supplementation with UDCA makes bile more hydrophilic and less cytotoxic, allowing a better flow through the bile ducts, preventing obstruction[33].
The routine use of UDCA in the management of CFLD is controversial. Results from a 2017 Cochrane review of UDCA for CFLD showed there was insufficient evidence to recommend its routine use in the management of CFLD[34]. However, Toledano et al. carried out a longitudinal population-based study involving >3,000 patients defined as having CFLD from the UK CF Registry between 2008 and 2013, results of which showed that treatment with UDCA was associated with prolonged overall survival (HR 0.70, 95% CI 0.52–0.96, P=0.026), specifically in those without cirrhosis (HR 0.50, 95% CI 0.36–0.69, P<0.0001) compared to those with cirrhotic disease (HR 1.19, 95% CI 0.46–3.10, P<0.71)[33].
Complications
Around 5–10% of people with CF who have CFLD develop cirrhosis and associated complications, such as portal hypertension, oesophageal varices and splenomegaly[30]. Therefore, annual monitoring with a hepatologist is recommended to screen and manage any complications that may arise[16].
Nutritional therapy
CFLD may lead to an increase in fat malabsorption and protein loss and requires careful management by a CF specialist dietitian to optimise calorific intake, especially with regards to protein. Supplementation of fat-soluble vitamins A, D, E and K should be given with serum monitoring, at least annually, to prevent deficiencies and toxicities. Pancreatic enzyme therapy should be optimised to ensure optimal absorption of triglycerides and essential fatty acids[25].
Monitoring
Most people with CF remain asymptomatic until pathological changes are pronounced, meaning that annual screening should be carried out in all people with CF ≥5 years of age to ensure an early diagnosis of CFLD[16]. Biochemical tests ( such as liver function tests, and albumin tests), clinical examination and imaging techniques (e.g., abdominal ultrasound) should be performed [see Figure 1][21]).
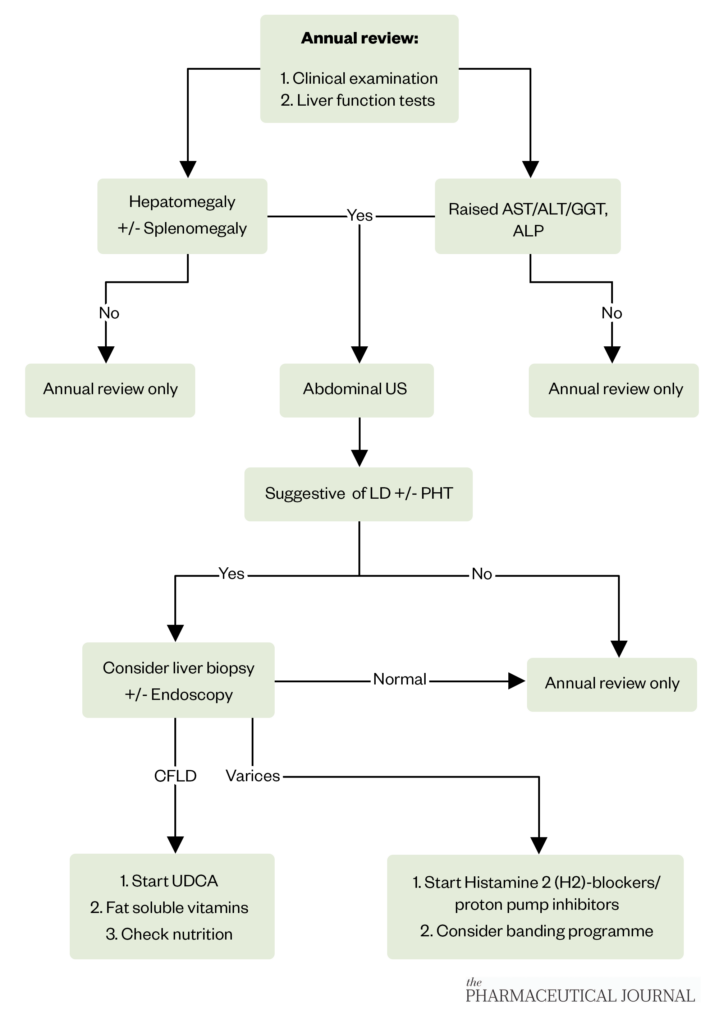
The Pharmaceutical Journal
CFTR modulators and CFLD
There is currently insufficient evidence to fully understand the impact the introduction of CFTR modulators have had on CFLD. However, Drummond et al. reported a sustained decrease in liver function tests (ALT, AST and GGT) following the initiation of lumafactor–ivacaftor in 28 people with CF aged over 12 years, suggesting a potential benefit of CFTR modulators in the management of CFLD[35].
It should be noted that CFTR modulators have been associated with rises in transaminase levels with a reported incidence of 10.9% in people with CF treated with ivacaftor/tezacaftor/elexacaftor, compared with 4.0% in the placebo group in registration studies[36]. Therefore, baseline measurement of transaminases (ALT or AST) and bilirubin is recommended before initiation of therapy, followed by regular monitoring (every three months) in the first year of treatment and at least annually after that.
Patients with underlying CFLD should be monitored more frequently. If liver function tests rise as follows; ALT or AST >5 x the upper limit of normal (ULN), or ALT or AST >3 x ULN with bilirubin >2 x ULN, treatment should be stopped, and liver function tests repeated at regular intervals until they return to baseline. Then the benefits and risks of restarting treatment should be considered. Treatment with CFTR modulators is not recommended in people with CF with severe liver disease and should only be used in moderate disease (Child-Pugh Class B) if there is a clear medical need and the benefit of treatment outweighs the risk of further hepatic insult. If the benefits outweigh the risk it should be used with caution at a reduced dose, consult the summary of product characteristics (SPC) for each product for further details[36].
Meconium ileus and distal intestinal obstruction syndrome
CFTR dysfunction leads to thickened, dehydrated mucus making the CF gut prone to obstruction. This manifests as meconium ileus in infants, and constipation and distal intestinal obstruction syndrome (DIOS) in older children and adults[37].
Meconium ileus is the obstruction of the intestine caused by thick adherent meconium that occurs in 15–20% of babies born with CF soon after birth[3,6]. Treatment depends on the severity of the obstruction with many infants requiring surgical intervention[9].
DIOS occurs when sticky mucus combines with thick faecal material, usually in the terminal ileum and proximal colon, resulting in complete or partial blockage[37]. It can be distinguished from constipation, defined as gradual faecal impaction of the total colon by radiological and clinical means[38].
The aim of treatment is removal of the complete or partial blockage with medication to avoid surgical intervention. Sodium meglumine diatrizoate, a hypertonic radio-opaque contrast medium, is administered orally or as an enema (by a specialist team only) to treat DIOS. It is a highly osmotic solution; therefore, patients should be adequately hydrated before administration to avoid hypovolemic shock (owing to rapid fluid shifting) and bowel perforation[37]. There is a significant risk of reoccurrence associated with a prior episodes of DIOS; therefore, implementation with a prophylactic regime, such as macrogol 3350 is recommended[37].
In 2010, Green et al., found differences between adult physician’s approaches to management of DIOS[39]. For incomplete DIOS, the three most common interventions were macrogol 3350, sodium meglumine diatrizoate and lifestyle modifications. While sodium meglumine diatrizoate, Klean-prep and non-surgical management (IV fluids, nasogastric aspiration and nil by mouth) were used to manage a complete blockage[39].
Management of constipation
Constipation, defined as the gradual impaction of the colon (see Table 2 for the European Society for Paediatric Gastroenterology, Hepatology and Nutrition Cystic Fibrosis Working Group’s definition), is very common in both paediatric and adult patients, affecting about 50% of patients per year[38,40]. However, there is currently no single strategy for the treatment of constipation in CF. A gradual escalation of treatments such as those used in people without CF is used. Osmotic laxatives, such as lactulose and macrogol 3350, are generally considered as first-line treatment for long-term use because they rehydrate the bowel[37]. Stimulant laxatives, such as senna and bisacodyl, and stool softeners, such as sodium docusate, have been used safely for short and long-term use in people with CF[37]. Supplementation with fibre and fibre-based laxatives (e.g. ispaghula husk) should be avoided in the people with CF because they may slow GI transit time and increase stool viscosity and could worsen symptoms[37].
N-acetylcysteine, a mucolytic, has been used as a treatment and in maintenance regimens in refractory constipation and DIOS. It breaks down the viscous mucus that is part of the mucofaeculant material that is adhered to the bowel wall[41]. The following dosing regimens have been used and are adjusted according to patient response:
- Children <12 years 100–200mg up to three times a day;
- Children >12 years and adults 200–400mg up to three times a day[42].
Other agents, such as prokinetic drugs that increase gastric motility (e.g. lubiprostone), metoclopramide and macrolide antibiotics, may need to be added to laxative regimes[43,44]. Lifestyle modifications, such as increased fluid or salt intake, assessment of pancreatic enzyme therapy and increased activity levels are also important[39].
Summary
While the majority of morbidity and mortality in CF is linked to respiratory health, non-respiratory manifestations can have a large impact on the lives of those with CF. They can feel like a further chronic illness on top of respiratory disease, which is already a burdensome illness and requires a significant amount of time and effort to maintain health. Furthermore, considering that all the manifestations discussed above can develop in the same person, it can been seen how significant polypharmacy can develop in people with CF. Therefore, pharmacists play a crucial role within the CF MDT in helping to initiate and educate on the many treatments required to maintain health in CF and assist people with CF in managing their significant treatment burden to make it as manageable as possible to promote lifelong adherence.
- 1Riordan JR. CFTR Function and Prospects for Therapy. Annu. Rev. Biochem. 2008;77:701–26. doi:10.1146/annurev.biochem.75.103004.142532
- 2Stoltz DA, Meyerholz DK, Welsh MJ. Origins of Cystic Fibrosis Lung Disease. N Engl J Med. 2015;372:351–62. doi:10.1056/nejmra1300109
- 3Wilschanski M, Durie PR. Pathology of pancreatic and intestinal disorders in cystic fibrosis. J R Soc Med. 1998;91:40–9. doi:10.1177/014107689809134s07
- 4Singh VK, Schwarzenberg SJ. Pancreatic insufficiency in Cystic Fibrosis. Journal of Cystic Fibrosis. 2017;16:S70–8. doi:10.1016/j.jcf.2017.06.011
- 5Nandhakumar N, Green MR. Interpretations: How to use faecal elastase testing. Archives of Disease in Childhood – Education and Practice. 2010;95:119–23. doi:10.1136/adc.2009.174359
- 6Gibson-Corley KN, Meyerholz DK, Engelhardt JF. Pancreatic pathophysiology in cystic fibrosis. J. Pathol. 2015;238:311–20. doi:10.1002/path.4634
- 7Lam KW, Leeds J. How to manage: patient with a low faecal elastase. Frontline Gastroenterol. 2019;12:67–73. doi:10.1136/flgastro-2018-101171
- 8Turck D, Braegger CP, Colombo C, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clinical Nutrition. 2016;35:557–77. doi:10.1016/j.clnu.2016.03.004
- 9Nutritional Management of Cystic Fibrosis. Cystic Fibrosis Trust. 2016.https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/Nutritional%20Management%20of%20cystic%20fibrosis%20Sep%2016.pdf (accessed May 2023).
- 10Pancreatin. British National Formulary. https://bnf.nice.org.uk/drugs/pancreatin (accessed May 2023).
- 11Patchell CJ, Desai M, Weller PH, et al. Creon® 10000 MinimicrospheresTM vs. Creon® 8000 microspheres—an open randomised crossover preference study. Journal of Cystic Fibrosis. 2002;1:287–91. doi:10.1016/s1569-1993(02)00103-0
- 12Beverley DW, Kelleher J, MacDonald A, et al. Comparison of four pancreatic extracts in cystic fibrosis. Archives of Disease in Childhood. 1987;62:564–8. doi:10.1136/adc.62.6.564
- 13Stead RJ, Skypala I, Hodson ME, et al. Enteric coated microspheres of pancreatin in the treatment of cystic fibrosis: comparison with a standard enteric coated preparation. Thorax. 1987;42:533–7. doi:10.1136/thx.42.7.533
- 14Pancreatic Enzymes Clinical Care Guidelines. Cystic Fibrosis Foundation. http://cff.org/medical-professionals/pancreatic-enzymes-clinical-care-guidelines (accessed May 2023).
- 15Freswick PN, Reid EK, Mascarenhas MR. Pancreatic Enzyme Replacement Therapy in Cystic Fibrosis. Nutrients. 2022;14:1341. doi:10.3390/nu14071341
- 16Debray D, Kelly D, Houwen R, et al. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. Journal of Cystic Fibrosis. 2011;10:S29–36. doi:10.1016/s1569-1993(11)60006-4
- 17Paravit-CF. Paravit-CF. https://www.paravit-cf.com (accessed May 2023).
- 18Granados A, Chan CL, Ode KL, et al. Cystic fibrosis related diabetes: Pathophysiology, screening and diagnosis. Journal of Cystic Fibrosis. 2019;18:S3–9. doi:10.1016/j.jcf.2019.08.016
- 19Management of cystic fibrosis diabetes. Cystic Fibrosis Trust. https://www.cysticfibrosis.org.uk/the-work-we-do/resources-for-cf-professionals/consensus-documents (accessed May 2023).
- 20Moran A, Dunitz J, Nathan B, et al. Cystic Fibrosis–Related Diabetes: Current Trends in Prevalence, Incidence, and Mortality. Diabetes Care. 2009;32:1626–31. doi:10.2337/dc09-0586
- 21De Boeck K, Weren M, Proesmans M, et al. Pancreatitis Among Patients With Cystic Fibrosis: Correlation With Pancreatic Status and Genotype. Pediatrics. 2005;115:e463–9. doi:10.1542/peds.2004-1764
- 22Ode KL, Chan CL, Granados A, et al. Cystic fibrosis related diabetes: Medical management. Journal of Cystic Fibrosis. 2019;18:S10–8. doi:10.1016/j.jcf.2019.08.003
- 23Onady GM, Stolfi A. Drug treatments for managing cystic fibrosis-related diabetes. Cochrane Database of Systematic Reviews. 2020;2020. doi:10.1002/14651858.cd004730.pub5
- 24Costa M, Potvin S, Hammana I, et al. Increased glucose excursion in cystic fibrosis and its association with a worse clinical status. Journal of Cystic Fibrosis. 2007;6:376–83. doi:10.1016/j.jcf.2007.02.005
- 25Staufer K. Current Treatment Options for Cystic Fibrosis-Related Liver Disease. IJMS. 2020;21:8586. doi:10.3390/ijms21228586
- 26Kinnman N, Lindblad A, Housset C, et al. Expression of cystic fibrosis transmembrane conductance regulator in liver tissue from patients with cystic fibrosis. Hepatology. 2000;32:334–40. doi:10.1053/jhep.2000.9111
- 27Lindblad A, Glaumann H, Strandvik B. Natural history of liver disease in cystic fibrosis. Hepatology. 1999;30:1151–8. doi:10.1002/hep.510300527
- 28Colombo C, Battezzati PM, Crosignani A, et al. Liver disease in cystic fibrosis: A prospective study on incidence, risk factors, and outcome. Hepatology. 2002;36:1374–82. doi:10.1002/hep.1840360613
- 29Toledano MB, Mukherjee SK, Howell J, et al. The emerging burden of liver disease in cystic fibrosis patients: A UK nationwide study. PLoS ONE. 2019;14:e0212779. doi:10.1371/journal.pone.0212779
- 30UK Cystic Fibrosis Registry Annual Data Report 2021. Cystic Fibrosis Trust. https://www.cysticfibrosis.org.uk/sites/default/files/2022-10/CFT_2021-Annual-Data-Report-WEB.pdf (accessed May 2023).
- 31Colombo C, Roda A, Roda E, et al. Evaluation of an oral ursodeoxycholic acid load in the assessment of bile acid malabsorption in cystic fibrosis. Digest Dis Sci. 1983;28:306–11. doi:10.1007/bf01324946
- 32Colombo C, Crosignani A, Assaisso M, et al. Ursodeoxycholic acid therapy in cystic fibrosis—associated liver disease: A dose-response study. Hepatology. 1992;16:924–30. doi:10.1002/hep.1840160412
- 33Paumgartner G. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525–31. doi:10.1053/jhep.2002.36088
- 34Cheng K, Ashby D, Smyth RL. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database of Systematic Reviews. 2017;2021. doi:10.1002/14651858.cd000222.pub4
- 35Drummond D, Dana J, Berteloot L, et al. Lumacaftor-ivacaftor effects on cystic fibrosis-related liver involvement in adolescents with homozygous F508 del-CFTR. Journal of Cystic Fibrosis. 2022;21:212–9. doi:10.1016/j.jcf.2021.07.018
- 36Kaftrio. European Medicines Agency . https://www.ema.europa.eu/en/medicines/human/EPAR/kaftrio (accessed May 2023).
- 37Abraham JM, Taylor CJ. Cystic Fibrosis & disorders of the large intestine: DIOS, constipation, and colorectal cancer. Journal of Cystic Fibrosis. 2017;16:S40–9. doi:10.1016/j.jcf.2017.06.013
- 38Houwen RH, van der Doef HP, Sermet I, et al. Defining DIOS and Constipation in Cystic Fibrosis With a Multicentre Study on the Incidence, Characteristics, and Treatment of DIOS. Journal of Pediatric Gastroenterology & Nutrition. 2010;50:38–42. doi:10.1097/mpg.0b013e3181a6e01d
- 39Green JA, Carroll WD, Gilchrist FJ. Comparing the management of constipation and distal intestinal obstruction syndrome between paediatricians and adult physicians. Journal of Cystic Fibrosis. 2018;17:e6–7. doi:10.1016/j.jcf.2017.09.007
- 40van der Doef HPJ, Kokke FTM, van der Ent CK, et al. Intestinal Obstruction Syndromes in Cystic Fibrosis: Meconium Ileus, Distal Intestinal Obstruction Syndrome, and Constipation. Curr Gastroenterol Rep. 2011;13:265–70. doi:10.1007/s11894-011-0185-9
- 41Green J, Carroll W, Gilchrist FJ. Interventions for treating distal intestinal obstruction syndrome (DIOS) in cystic fibrosis. Cochrane Database of Systematic Reviews. 2018;2018. doi:10.1002/14651858.cd012798.pub2
- 42Clinical guidelines: Care of children with cystic fibrosis, 2023. Royal Brompton and Harefield Hospitals, Guy’s and St Thomas’s NHS Foundation Trust . www.rbht.nhs.uk/childrencf (accessed May 2023).
- 43O’Brien CE, Anderson PJ, Stowe CD. Lubiprostone for Constipation in Adults with Cystic Fibrosis: A Pilot Study. Ann Pharmacother. 2011;45:1061–6. doi:10.1345/aph.1q219
- 44Longo WE, Vernava AM. Prokinetic agents for lower gastrointestinal motility disorders. Diseases of the Colon & Rectum. 1993;36:696–708. doi:10.1007/bf02238599