Key points:
- Advances continue to be made in the development of precision medicines for cystic fibrosis (CF);
- To date, two genotype-specific medicines – ivacaftor (Kalydeco®) and ivacaftor/lumacaftor (Orkambi®) – have been launched in the EU;
- For the majority of people living with CF today, ‘conventional’ drug treatments (e.g. mucus modifiers and inhaled antibiotics) will still be required to reduce symptoms and gain stability because newer agents cannot reverse existing damage to the airways caused by the disease processes;
- The financial impact of new drugs that correct the basic defect in CF or those that can reduce symptoms could become a barrier to access and, ultimately, development;
- There should be a focus on supporting adherence to treatments to ensure people with CF gain maximal benefit from their medicines.
Introduction
Cystic fibrosis (CF) is a life-limiting, inherited condition, with a reported incidence of 1 in 3,000 live births in northern Europe[1]
. In the UK, there are now more than 10,000 people living with the condition, with an estimated 100,000 people affected globally[2],[3]
. Previously a disease with high childhood mortality, the number of adults living with CF is ever increasing and now accounts for 60% of the CF population in the UK. In 2015, the median predicted survival was 45.1 years and median age at death was 28.0 years for people with CF in the UK[2]
.
With the dawn of precision medicines, an appreciation of genetics and pharmacogenomics is becoming increasingly important for pharmacists. An understanding of the basic defect and differences conferred by different genotypes is required to understand the mechanism of action of precision medicines and medicines that target the consequences of the basic defect. People with CF are also encouraged to know their genotype and how this determines the treatments available to them. Following a drive to ensure all those affected have their genotype documented, more than 98% of people with CF in the UK have been genotyped[2]
.
The basic defect
CF is an autosomal recessive disease, caused by a mutation in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein, a channel that transports ions through the apical membrane of epithelial cells[4]
. Some 2,000 CFTR mutations have been identified to date, although fewer than 10% of these are thought to be disease-causing[5]
. Six classes of CFTR mutation have been described, which, in general, lead to complete absence, reduced expression or expression of dysfunctional CFTR protein at the cell surface; these give rise to different phenotypic expressions of the disease[6]
(see ‘Table 1: Cystic fibrosis genotypes – UK distribution’). The different classes of mutations have been associated with significant differences in survival and median age at death, although probability of long-term survival may also be determined by several factors independent of CFTR function (e.g. gene modifiers and age at diagnosis)[7],[8]
.
The most common mutation is a three base pair deletion, resulting in the omission of a phenylalanine residue at position 508 of the CFTR protein, denoted by F508del. Around 90% of people with CF in the UK express this mutation on at least one allele[2]
.
| Table 1: Cystic fibrosis genotypes – UK distribution | ||||||
|---|---|---|---|---|---|---|
| Mutation Class | I | II | III | IV | V | VI |
| Genotypes with UK prevalence >1% (protein/legacy name [%])[2] | p.Gly542X (3.45%) 621+1G->T (2.21%) 1717- 1G- >A (1.24%) 1898+ 1G- >A (1.17%) | p.F508del (90.49%) p.Asn1303Lys (1.41%) p.Ile507del (1.04%) | p.G551D (5.67%) | p.R117H (4.61%) | None | None |
| Functional consequence[4] | Few to no mature CFTR proteins are formed | Defective processing and transport – reduced quantity of CFTR protein delivered to cell surface | CFTR delivered to cell surface but reduction in channel-open probability | CFTR at the cell surface but impaired movement of ions through channel | Decreased quantity of CFTR protein at cell surface | Accelerated turnover of CFTR protein at the cell surface reduces quantity |
| Precision medicine target/examples | Ataluren | Ivacaftor + lumacaftor | Ivacaftor | Ivacaftor | Potentially gene therapy | Potentially gene therapy |
Sources and selection criteria
To identify material for this narrative review, searches were performed on PubMed/MEDLINE using combinations of the search terms ‘cystic fibrosis’, ‘cystic fibrosis/drug therapy’, ‘cystic fibrosis transmembrane conductance regulator’, ‘ivacaftor’, ‘lumacaftor’, ‘ataluren’, ‘personalised medicine’ and ‘precision medicine.’ The search was first limited to English language articles concerning human subjects published in the past five years. The reference lists of relevant articles were also reviewed, and articles and documents of particular interest retrieved.
Pathophysiology
The CFTR protein is expressed in the epithelial cells of many organs, including, but not limited to, the airways, pancreas, gastrointestinal tract and sweat glands[9]
. This distribution accounts for the pathophysiology of the disease and key targets for drug development. CF is most often characterised by elevated sweat chloride, pancreatic insufficiency, and inflammation and infection of the airways.
Chronic infection and its sequelae result in progressive decline in pulmonary function and pulmonary failure; the most common cause of morbidity and mortality in those affected[10]
. In the CF lung, impaired CFTR functioning leads to impaired chloride and bicarbonate ion transport, and enhanced sodium absorption across the airway epithelial cells[10]
. This shift in electrolytes causes an increase in water absorption from the epithelial surface, which leads to more viscid (sticky) mucus on the surface of the airways. This mucus accumulates as a result of reduced clearance and, with the ciliary elevator compromised, further stasis of mucus occurs, resulting in a cycle of recurrent infections and inflammation[11]
. This, in turn, weakens the lung defences and causes tissue destruction (bronchiectasis), which leads to a decline in lung function, most commonly demonstrated by loss of forced expiratory volume in the first second of expiration (FEV1)[12]
. Traditionally, these downstream effects in the lungs have been the targets for drugs aimed at reducing infection (inhaled antibiotics) and changing the characteristics of the mucus (mucus modifiers).
Other common complications include pancreatic exocrine and endocrine insufficiency, malnutrition, CF-related liver disease, gastrointestinal disorders (e.g. distal intestinal obstruction syndrome [DIOS]), reduced bone mineral density and male infertility. As might be expected from a disease affecting so many different systems, polypharmacy in CF is common and has been associated with poor adherence to treatments[13]
.
Therapeutic options
Management of CF is tailored to the individual and their comorbidities. The mainstay of treatment for lung disease is the use of airway clearance techniques (ACT) to expectorate (eject and remove) mucus, often with adjuncts, such as the mucus-modifying drugs recombinant human dornase alfa (rhDNase), hypertonic saline and mannitol[14]
. Chronic infection is suppressed with oral or inhaled antibiotics, primarily targeting Pseudomonas aeruginosa infection, while infective exacerbations are treated with oral or intravenous antibiotics depending on severity[15]
. A spectrum of emergent fungal diseases may require treatment with antifungals and oral corticosteroids; and inflammation may be dampened with azithromycin. Exocrine pancreatic insufficiency is treated with pancreatic enzyme replacement therapy (PERT) and other nutritional support, while endocrine insufficiency, ultimately manifesting as CF-related diabetes (CFRD), is treated with insulins[14]
. Fat-soluble vitamins often require supplementation to ensure optimal vitamin levels and prevent hypovitaminosis A, D, E and K, which each have their own complications[14]
.
While the treatment strategy for people with CF may employ several of these therapeutic options, the basic genetic defect and pulmonary disease have been the prime targets for drug development over the past decade. These developments are highlighted below.
‘Conventional’ pulmonary treatments
Table 2 provides a brief overview of the most notable drugs specifically developed for the management of pulmonary complications of CF over the past 20 years, although the past decade has seen the greatest number of drug developments. These drugs broadly fall into two categories: mucus modifying agents and inhaled antibiotics. The aim of therapy is to stabilise or prevent decline in lung function (FEV1, which is measured in absolute terms [volume, in millilitres (mL)] and in relative terms [% predicted]), and reduce or prevent the occurrence of acute pulmonary exacerbations that have negative consequences in terms of immediate morbidity, as well as long-term morbidity and mortality[16]
.
| Table 2: Selection of ‘conventional’ treatments that target pulmonary disease in cystic fibrosis approved in the EU | |||
|---|---|---|---|
| Drug | Date of first marketing authorisation | Recommended dosing | Place in therapy (UK) |
| Dornase alfa (DNase®, Roche Products Ltd)[75] | January 1994 | Child >5 years and adult: 2.5mg by inhalation OD. Some individuals aged over 21 years may benefit from BD dosage | Introducing rhDNase early in the disease may be beneficial to improve pulmonary function |
| Tobramycin solution for inhalation (TSI)[76] | September 2006 | Child >6 years and adult: 300mg BD for 28 days, followed by 28 days without inhaled tobramycin (i.e. alternate months) | For eradication or suppression of chronic Pseudomonas aeruingosa |
| Tobramycin inhalation powder (TIP) (TobiR) Podhaler, Novartis Pharmaceuticals UK Ltd.)[77] | July 2011 | Child >6 years and adult: 112mg (four capsules) BD for 28 days, followed by 28 days without inhaled tobramycin (i.e. alternate months) | Alternative to TSI in those to whom TSI would otherwise have been considered |
| Aztreonam lysine (Cayston®, Gilead Sciences Ltd)[78] | September 2009 | Child >6 years and adult: 75mg TDS for 28 days, followed by 28 days without inhaled aztreonam (i.e. alternate months) | Suppression of chronic P. aeruginosa in those in those who have failed to tolerate or continue to decline despite use of colistimethate sodium/tobramycin |
| Colistimethate sodium (Colobreathe®, Forest Laboratories UK Ltd)[79] | February 2012 | Child >6 years and adult: 1,662,500 units (125mg) BD | Suppression of chronic P. aeruginosa in those who do not tolerate or struggle to adhere with nebulised colistimethate |
| Mannitol (Bronchitol®, Pharmaxis Pharmaceuticals Ltd)[80] | April 2012 | Adult: 400mg BD | Option for improving sputum clearance in those who cannot use rhDNase owing to ineligibility, intolerance or inadequate response to rhDNase, when lung function is rapidly declining and other osmotic agents are not considered appropriate |
| Levofloxacin (Quinsair®, Raptor Pharmaceuticals Europe B.V.)[81] | March 2015 | Adult: 240mg BD | Suppression of chronic P. aeruginosa – place in therapy in relation to other inhaled antibiotics to be established |
| Hypertonic saline[82] | Not applicable | Child and adult: 4mL of 3–7% solution (as tolerated) BD | Option for improving sputum clearance in those intolerant of, or fail to benefit from rhDNase (either alone or in combination) |
Mucus modifying agents
Owing to abnormalities in ion transport within the airway epithelial cells and subsequent depletion of the airway surface liquid, the clearance of airway secretions in CF is impaired[11]
.
rhDNase has been extensively investigated in CF and is now considered to be part of the current standard of care for people with CF, owing to its role in improving lung function and reducing acute pulmonary exacerbations[17]
. Recently, greater attention has turned to timing and method of administration of rhDNase. A Cochrane review concluded that timing can “be largely based on pragmatic reasons or individual preference”[18]
.
In the 1990s, advances were made in the application of inhaled hypertonic saline as a means to rehydrate the airway surface and improve rheological properties of sputum to aid clearance. This was found to confer benefits by improving lung function and reducing the number of exacerbations[19]
. Since then, hypertonic saline has been embedded into routine practice. Recently, there has been a suggestion that hypertonic saline use during an acute pulmonary exacerbation may also expedite symptom resolution[20]
. Early studies suggest a dose-dependent effect of hypertonic saline and although there is debate regarding the optimal concentration of hypertonic saline, in practice, this is largely driven by commercial availability and individual tolerance[21],[22]
.
One of the more recent advances in mucus modifiers has been the development of inhaled mannitol – a sugar alcohol that may improve airway surface hydration in CF by creating an osmotic gradient in the airway lumen. Significant improvements in FEV1 were observed over 26 weeks in two large phase III randomised controlled trials (RCTs) in subjects with mild-to-severe pulmonary impairment when mannitol was added to current treatment, despite higher treatment-related adverse effects in the mannitol group[23],[24]
.
A recent pooled analysis of the two mannitol RCTs found that both the mean absolute change in FEV1 (73.42mL) and relative change in FEV1 (3.56% predicted) from baseline were statistically significant and clinically meaningful in the mannitol group versus control[25]
. The observed improvement in FEV1 was independent of rhDNase use and was accompanied by a 29% reduction in the incidence of pulmonary exacerbations[25]
. Importantly, improvements were observed in adults but not in children or adolescents, which is in keeping with the marketing authorisation that excludes the treatment of children[25]
.
In the UK, rhDNase or hypertonic saline should be considered for all individuals with CF aged over six years as an adjunct to airway clearance[14]
. Within the NHS in England, a commissioning policy sets criteria for prescribing inhaled therapies for people with CF, driving equity in access to effective treatments that are deemed to be affordable within the NHS[26]
. This guidance sets out the recommendations for inhaled mucolytics – rhDNase and mannitol. rhDNase is recommended in patients aged six years and over to prevent mucus plugging and deterioration in lung function; however, it is also permitted in certain instances in children aged under six years to prevent decline and reduce the requirement for intravenous antibiotics for acute pulmonary exacerbations.
The benefits of hypertonic saline are independent of treatment with rhDNase, therefore, it may be used concomitantly in those who benefit from rhDNase yet require additional adjuncts to ACT, or as an alternative when rhDNase is not tolerated or is not effective[27]
.
Mannitol – considered a third-line agent – is recommended by the National Institute for Health and Care Excellence (NICE), England’s health technology assessment body, as an option for treating CF in adults who cannot use rhDNase because of ineligibility, intolerance or inadequate response; in those with rapidly declining lung function; and for whom other osmotic agents (e.g. hypertonic saline) are not considered appropriate[28]
.
Inhaled antibiotics
P. aeruginosa is one of the most significant pathogens to affect the airways of people with CF[15]
. Strategies exist to prevent chronic infection by aggressively treating new isolates to eradicate the organism from the airways and to suppress chronic infection, which leads to a damaging inflammatory response with associated decline in pulmonary function[15]
.
The optimal antibiotic eradication therapy for P. aeruginosa has not yet been determined; a recent Cochrane review found evidence of benefit with both colistimethate sodium and tobramycin[29]
. Eradication remains a main focus of investigations, highlighted by a recent study that found that a 28-day course of aztreonam lysine might be an alternative option for eradication of new onset P. aeruginosa in children, with eradication rates consistent with other inhaled antibiotics studied for eradication[30]
.
The likely outcome in those who have previously acquired P. aeruginosa is chronic infection, which is defined as individuals in whom more than 50% of airway cultures have been culture positive for P. aeruginosa in the preceding 12 months[31]
. There is an increased association of chronic P. aeruginosa with age; recent data suggest that almost 60% of adults aged 32–35 years in the UK are chronically infected[2]
.
The relative efficacies of inhaled antibiotics for suppression of chronic pseudomonal infections in CF have been compared in a network meta-analysis[32]
. Following analysis of seven RCTs, it was determined that all studied antibiotics (tobramycin, including tobramycin solution for inhalation [TSI] and tobramycin inhalation powder [TIP]; colistimethate sodium; and aztreonam lysine) have comparable efficacies for the treatment of chronic P. aeruginosa, measured by change in FEV1 from baseline, and that the change from baseline was clinically meaningful over the study period (i.e. an improvement of at least 3.5% predicted)[32]
.
Since the analysis was conducted, two further inhaled antibiotics have been launched in the UK: colistimethate sodium dry powder inhaler (CDPI) and levofloxacin inhalation solution (LIS). When compared with three 28-day on-off cycles of TSI in a phase III trial of patients with stable CF ≥6 years with chronic P. aeruginosa infection, CDPI was found to be non-inferior to TSI in the primary end point (change in mean FEV1 percentage predicted from baseline to week 24)[33]
. Non-inferiority was also demonstrated with LIS when compared with TSI over three 28-day on-off cycles in a phase III trial in patients with CF ≥12 years with chronic P. aeruginosa infection previously treated with TSI[34]
. LIS was well tolerated and met the primary end point (relative change in FEV1 percentage predicted) over a 28-day cycle[34]
.
With an increasing array of inhaled antibiotics comes the challenge of deciding which to use when and which combination. Most studies have compared single agents against another established agent in an intermittent fashion, whereas in practice, individuals may be prescribed continuous inhaled antibiotic therapy, usually alternating monthly between agents. The comparison of different regimens has been poorly described in the literature.
In the UK, the consensus view is that 90% of patients chronically infected with P. aeruginosa should be prescribed at least one inhaled antibiotic[2]
. This is executed through NHS guidance for CF: eradication of P. aeruginosa may be attempted with colistimethate sodium or tobramcycin, with a stepwise approach recommended for management of chronic P. aeruginosa infection[26]
. In general, colistimethate sodium is the first-line treatment, tobramycin is the second-line treatment and aztreonam lysine is the third-line treatment. Progression through these agents is determined by tolerability and effectiveness, which for inhaled antibiotics is defined by stability in FEV1 and absence of acute pulmonary exacerbations[26]
. NICE guidance on the use of dry powder colistimethate sodium and tobramycin preparations exists, although this guidance has largely been incorporated into NHS commissioning policy, widening access to those who struggle to adhere to liquid formulations, as well as those who are intolerant[26]
.
Ultimately, antibiotic regimens for chronic infection will be tailored to the individual based on their response and preferences. The place of LIS has not yet been determined, although an update to the NHS policy is expected shortly.
Precision medicines
While gene therapy trials in CF continue to make progress (an account of which has recently been published in The Pharmaceutical Journal
[35]
), drugs designed to restore CFTR function have risen in prominence in recent years, both in clinical and political spheres. To date, two drugs have reached market, with several others currently under investigation[36]
. Table 1 shows each class of mutation and its different characteristics. The most effective treatments to date have exploited a particular characteristic of the dysfunctional cellular process. ‘Figure 1: Treating a population with precision medicines – what could that look like in the near future’ shows the proportion of the CF population in the UK who may derive benefit from each of the transformative therapies, either marketed or in development, based on class of mutation. The first three classes are discussed in greater detail because these are the current priorities for drug developments.
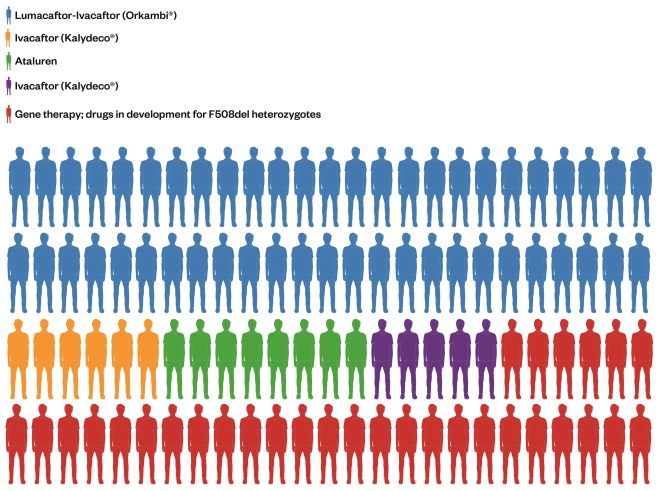
Figure 1: Treating a population with precision medicines – what that could look like
Pictorial breakdown of how the UK cystic fibroris population may be treated in the future. Each figure represents 1% of the UK CF population based on their genotypes.
Class I mutations
Class I nonsense mutations result in an absence of functional CFTR protein owing to the presence of a premature termination codon (PTC) within the messenger RNA (mRNA). Drugs that promote ribosomal read-through of stop codons, permitting complete translation of mRNA, may represent a potential treatment strategy for this type of defect[4]
.
Ataluren (Translarna®, PTC Therapeutics Ltd.; New Jersey, United States) – an orally administered, small-molecule compound recently approved for use in another genetic disorder caused by a nonsense mutation (Duchenne muscular dystrophy) – promotes ribosomal read-through of the PTC and has been demonstrated to produce full length, functional CFTR[37],[38]
.
A phase III trial designed to evaluate the effect of ataluren in subjects aged ≥6 years with CF caused by a nonsense mutation failed to meet its primary end point (relative change in percentage predicted FEV1 at week 48) and there was no statistically significant difference in rates of exacerbation between the treated and placebo-controlled groups[39]
. Aminoglycosides have previously demonstrated to promote read-through of PTCs, therefore, a post-hoc analysis in subjects not concomitantly receiving inhaled tobramycin was undertaken. A modest increase in FEV1 over the treatment period (5.7% predicted) and corresponding decrease in rate of exacerbations (by 40%) in the treatment group were observed, both of which were statistically significant[39],[40]
.
A further phase III study is currently under way that will evaluate the safety and efficacy of ataluren in subjects with a class I ‘nonsense’ mutation who are not receiving chronic inhaled aminoglycosides[41]
. If the results generated by the earlier post-hoc analysis are confirmed in this trial, more than 600 people with CF in the UK could stand to benefit from ataluren[42]
.
Class II mutations
As class II mutations are by far the most common, they represent a prime target for precision medicine in CF. In the UK, F508del homozygotes account for 50% of the CF population, with a further 40% being heterozygotes[2]
. Class II mutations result in a misfolded protein that is destroyed by the cell machinery. Drug targets are required to correct the processing of CFTR and are therefore termed ‘CFTR correctors’.
The F508del mutation results in severely reduced expression of the CFTR protein at the epithelial membrane; those channels that do reach the cell surface exhibit disrupted opening. A two-pronged approach to ameliorate the effect of this mutation has, therefore, been investigated: a CFTR corrector to increase the amount of functional mutated CFTR, combined with a CFTR potentiator to increase the channel opening probability[43]
.
Lumacaftor – the first-in-class CFTR corrector – in combination with ivacaftor (Orkambi®, Vertex Pharmaceuticals [Europe] Ltd; UK) was granted a marketing authorisation in the EU in November 2015, following evaluation in two phase III placebo-controlled studies in subjects with CF who were homozygous for the F508del mutation[44]
. In both studies there was a statistically significant but modest improvement in the change in FEV1 percentage predicted from baseline at week 24 (mean relative treatment difference 4.3 to 6.7%). While in itself, this discreet change in absolute FEV1 percentage predicted is unlikely to be clinically meaningful, this could represent stability in pulmonary function – the outcomes from longer studies to confirm this effect are eagerly anticipated. More notably, a statistically significant reduction in the rate of pulmonary exacerbations was observed in pooled analyses (between 30% and 39% in the treated groups), with a corresponding trend towards fewer hospital admissions and fewer courses of intravenous antibiotics in the treated groups[44]
. This is particularly important because fewer exacerbations would be expected to confer better long-term outcomes.
Another CFTR corrector, VX-661, is currently being investigated in phase III studies in combination with ivacaftor in F508del homozygotes, and two further ‘next generation’ correctors are in clinical development (VX-152 and VX-440) with phase II studies of triple therapy (VX-152/VX-661/ivacaftor and VX-440/VX-661/ivacaftor) planned to commence in late 2016[45],[46]
.
Class III mutations
Class III mutations result in expression of impaired CFTR channels at the cell membrane, such that the probability of the channel being open (and functioning normally) is reduced.
Ivacaftor (Kalydeco®, Vertex Pharmaceuticals (Europe) Ltd; UK) was the first breakthrough drug designed to target the basic CFTR defect. Granted a marketing authorisation in July 2012, ivacaftor is a first-in-class CFTR potentiator that acts by increasing the flow of ions through activated CFTR channels by increasing the open probability of the CFTR channel once it has reached the apical cell membrane[47]
.
The efficacy of ivacaftor has been demonstrated in several studies. In the seminal phase III study conducted in subjects ≥12 years with CF and at least one copy of the G551D mutation, a statistically significant and clinically meaningful increase in FEV1 was observed in the ivacaftor group (10.6% predicted) compared with the placebo group[48]
. This improvement in FEV1 was observed after only two weeks of treatment, and was sustained for the 48-week duration of the study. A statistically and clinically significant reduction in rate of pulmonary exacerbations was also observed in the treated group (55% risk reduction) and treated subjects gained an average of 2.7kg over the course of the study. Of particular note was the statistically significant reduction in sweat chloride in the treated group to a mean of 47.8mmol/L, making ivacaftor the first agent to confer a reduction in sweat chloride to a value below the usual diagnostic threshold for CF.
The outcome of this study was arguably the most significant milestone in the management of CF since the identification of the responsible gene: for the first time it was demonstrated that CFTR function could be restored and some of the damaging effects of the disease could be reversed.
Similarly encouraging results were also observed in children aged 6–11 years with the same mutation, and in individuals aged ≥6 years with selected other ‘gating’ (class III) mutations[49],[50]
. Safety and pharmacokinetics have been studied in children aged 2–5 years, and with exposure to the drug similar to that previously reported, there is every reason to believe ivacaftor will improve the trajectory of disease progression in this age group[51]
.
Real-life data suggest similar results are being seen in those treated with the drug in practice, with similarly depressed sweat chloride and improvements in FEV1 and a low rate of discontinuation[2]
.
The ability to depress sweat chloride has since been replicated in phase II studies of a new investigational potentiator, QBW251, which is being developed by Novartis Pharmaceuticals (Switzerland).
Class IV–VI mutations
R117H, a class IV mutation, is an example of a residual function mutation associated with milder clinical manifestation and increased survival, for which ivacaftor has also been evaluated[52]
. While the primary end point (absolute change from baseline FEV1 percentage predicted over the study period) was not met in this trial, improvements were observed in sweat chloride and quality of life outcomes, and a pre-specified subgroup analyses in subjects aged ≥18 years found a statistically significant (but clinically modest) improvement in FEV1[53]
.
With limited investigation into precision medicines for class IV to VI mutations, these may represent particular targets for gene therapy in the future.
New challenges
While advances in precision medicines have the potential to improve outcomes in CF, several challenges also arise. With high costs, they are already being scrutinised for affordability. They do not necessarily resolve difficulties with adherence to treatments and, as yet, relatively little is known about their long-term effects in people who will (in most cases) take these treatments for many years. The role of the pharmacist in the management of CF has recently been described and central to the role is an understanding of the current challenges in treating CF[54]
.
Funding and cost-effectiveness
There is an increasing public expectation that all treatments will be made universally available, irrespective of cost. However, the financial challenge to treating CF in the UK and elsewhere currently seems insurmountable, and is only set to worsen. The reasons for the challenges are complex and include increasing survival and the associated increase in comorbidities; an increasing population – latest forecasts indicate an increase in the CF population by around 50% by 2025 in western Europe (20% in the child population and 75% in the adult population); and high acquisition costs associated with precision medicines[55]
. This ‘perfect financial storm’ comes at a time when NHS spending is under increasing scrutiny, with spending on specialised services in England (including CF care) having increased materially by 6.3% each year between 2013–2014 and 2015–2016.
In the UK and elsewhere in Europe, there is good access to ‘conventional’ treatments for CF (e.g. colistimethate sodium, tobramycin and rhDNase). However, access to newer formulations of inhaled therapies and precision medicines is not standardised throughout the EU[56]
. In the UK, access is generally equitable, however, there can be long delays between a medicine being granted a marketing authorisation and being authorised for routine use from central funds.
Despite being the first drug reported to be developed through venture philanthropy, ivacaftor is currently one of the world’s most expensive treatments. In 2013, following a positive NHS National Institute for Health Research (NIHR) Technology Assessment Report, ivacaftor reached CF clinics in the UK, with an NHS indicative price of £182,000 per person per year[57]
. With 439 individuals reported to have been prescribed the treatment between June 2012 and December 2015, the annual bill to the NHS for ivacaftor was £55m in 2015–2016 (a 50% increase on the previously quoted £110m annual budget for CF) and the bill for ivacaftor is set to rise to around £60m in 2016–2017[2],[57],[58]
. Interestingly, although the health technology assessment (HTA) for ivacaftor concluded that it was an effective treatment, the authors determined that there was “no clear benchmark to indicate whether or not ivacaftor should be considered cost-effective”[59]
. In this instance, a discount was agreed between the NHS and the manufacturer to improve cost-effectiveness and make it available for prescribing[60]
.
On 27 July 2016, NICE, who were tasked with determining the cost-effectiveness of the lumacaftor–ivacaftor combination (Orkambi®), published a recommendation that it would not be recommended for use in the NHS, a position already adopted by the Scottish Medicines Consortium (SMC)[61],[62]
. Despite a lower NHS indicative price of £104,000 per person per year, it could cost around £500m per year to treat all the eligible individuals in the UK with Orkambi®[2],[61]
. It was determined that this considerable cost of Orkambi® was not justified compared with the current standard of care[61]
. Whether or not an agreement can be reached over a price that could be deemed affordable to the NHS is yet to be seen.
Longer term evaluation of these agents will be required to determine their true ability to modify the course of the disease over many years and indeed long-term cost-effectiveness because the current expectation is that these agents could be prescribed life-long[36]
.
Clinical trials and affordability of drugs – ethical considerations
Precision medicines, by virtue of high development costs and relatively limited reach when marketed, may attract exorbitant prices. Given the high cost for such a modest benefit, the negative NICE opinion on the cost-effectiveness of Orkambi® may not seem extraordinary, but it should be viewed as a cautionary tale.
The concept of distributive justice suggests that risks of research cannot be assumed by one cohort, if the cohort is unlikely to benefit from that research. The Declaration of Helsinki, the ethical framework for medical research involving human subjects, asserts that risk assessment prior to involving human subjects in research must compare the risks and burdens to the individuals and groups involved, with the foreseeable benefits to them and others affected by the condition[63]
.
Previously, this principle seemed most relevant to those wishing to study the effects of drugs in subjects in developing nations, whose governments may not be able to afford an intervention after it has been tested in their populations. However, with the advent of precision medicine, the cost of providing expensive interventions to a population post-trial and its affordability by healthcare purchasers is now a universal consideration. As research should be responsive to the health needs of the host country, post-trial obligations must now be given greater emphasis, not least in CF. In the current financial climate of the NHS, researchers, trial sponsors, commissioners and policymakers must consider (from an early time point in a drug’s development journey) how any benefits demonstrated in a clinical trial may be made available to the wider population.
Studies of Orkambi®, for example, were conducted in the UK, however, the pricing structure and subsequent decisions not to approve it for use in the UK on the basis of cost-effectiveness represents a failure to properly address the challenge of justice in clinical trials. It is hoped that the Department of Health’s ‘Accelerated access review’ will help to strengthen the relationship between the NHS and innovators in the future, and lead to better access by managing the introduction of innovative technology as soon as possible, where evidence of longer-term value has not yet been established[64]
.
This issue may also be helped by the recent recommendation to the Committee for Medicinal Products for Human Use (CHMP) to revise the current guideline on the clinical development of medicinal products for the treatment of CF, in light of current and anticipated future requirements for effective trial design in CF[65]
. More targeted trial design in CF may permit more robust analysis of cost-effectiveness of new drugs and support policymakers to determine affordability.
As interest in developing precision medicines for CF continues to grow and more treatment options reach market, some of the issues relating to cost may, in part, be resolved by a more competitive marketplace.
Adherence to treatments
A well described challenge in patients with CF who are often prescribed a multitude of treatments to ameliorate the disease is adherence to treatments. The reasons for reduced adherence are complex, but are likely to include a combination of factors, which may include lack of time to be fully adherent with a complex regimen, depression and anxiety, and a preference not to take treatments in public (e.g. insulin, inhaled treatments or pancreatic enzyme replacements). Supporting patients to improve adherence is equally complex because no specific intervention has been found to be universally beneficial. Instead, a person-centred approach must be taken to tailor any interventions adopted, which may include prioritisation of treatments based on what the individual perceives to be most important, and employing behavioural strategies[66]
.
One large retrospective study conducted with records of patients with CF in the United States – using medication possession ratio as a proxy for adherence – found that around half of the sampled cohort filled less than half of their prescriptions, which is consistent with known levels of adherence in other chronic diseases[67]
. In that cohort, thought to be representative of the general CF population, average drug-specific adherence rates ranged from 40% to 57%. Overall mean adherence to pulmonary treatments was 48%, and appeared to be greater in younger subjects and those with a higher treatment complexity, which perhaps reflects attention to adherence in those with more advanced disease.
It has been demonstrated that there is an association between lower medication adherence and the need for intravenous antibiotics to treat pulmonary exacerbations[68]
. In the same study, the investigators also observed preservation of lung function over a 12-month period in those subjects with adherence rates >80% compared with those with <80%, who experienced a loss in lung function. Although not statistically significant, this is a reasonably predictable consequence of lower adherence, when it is considered that increasing survival rates are thought to be caused, in part, by advances in treatments available. Lower adherence to treatments in CF has also been associated with increased healthcare costs[69]
.
As it has been demonstrated that lower rates of adherence are associated with worse outcomes for individuals and increasing costs, it is possible that targeted adherence support could improve these indices. While pharmacists are well placed to support adherence monitoring, achieving this is difficult in practice, with more than three-quarters of CF specialist pharmacists in a survey describing inadequate adherence monitoring, associated with limited opportunities for sufficient specialist pharmacist involvement[70]
.
Despite the high cost of ivacaftor, surprisingly few studies have established adherence rates to ivacaftor in practice. A small study conducted by a UK investigator using electronic monitoring found that even adherence rates to ivacaftor are suboptimal and, at 61%, is comparable with other therapies used in the management of CF. Whereas, a team from the United States determined that fewer than three-quarters of patients taking ivacaftor had a medicines possession ratio >0.8[71],[72]
. If these data are to be believed representative, it suggests a significant problem, not least for those who may not gain the full benefits of treatment with ivacaftor, but also for the wider health economy because the decision to fund treatment was based on cost-effectiveness calculations that assumed 91% adherence based on trial data[48]
. This reduction in adherence would be likely to confer a reduction in efficacy outcomes.
Complexity in treatment regimens
With increased survival, several new issues in the management of CF are emerging. In an ageing population, common age-related conditions, such as hypertension, hyperlipidaemia and associated risk of cardiac disease, and malignancies are becoming new therapeutic challenges. With increasing longevity also comes an increasing lifetime exposure to drugs, some with cumulative adverse effects; the risk of hearing disturbances and renal impairment observed with aminoglycosides, for example, is greater with higher exposure. For many drugs, such as the newer agents described herein, any risks associated with long-term treatment have yet to be established.
Increasing age and comorbidities often increase the complexity of the treatment regimen and although some evidence suggests that increasing complexity does not necessarily translate into reduced adherence, there are other important issues, such as management of drug interactions and adverse drug reactions. Lumacaftor is a strong inducer of CYP3A, while on its own, ivacaftor (a substrate of CYP3A4 and CYP3A5) is a weak inhibitor of CYP3A[73]
. Those treated with either drug should avoid concomitant treatment with strong CYP3A inducers (e.g. rifampicin), while dose adjustments of ivacaftor may be required for those on treatment with moderate to strong inhibitors of CYP3A (e.g. itraconazole and clarithromycin). Lumacaftor may substantially decrease hormonal contraceptive exposure, therefore, alternative approaches should be considered when starting treatment with lumacaftor/ivacaftor.
Increasingly, consideration must be given to managing treatments pre-conception and during pregnancy. While some drugs used in the management of CF are supported by a degree of reproductive safety data, little is currently known about the effect of newer agents during pregnancy. As outcomes in pregnancy have been established to be closely related to lung function and stability of lung disease, a balance must be struck between the foetal and maternal risks and benefits to continuing or suspending drug treatments[74]
.
Complex regimens – particularly those including genotype-specific medicines or drugs used in special populations (e.g. those who wish to become pregnant or those with renal or hepatic dysfunction) – should be prescribed in collaboration with a CF-specialist pharmacist.
Conclusion
While clinical application of gene therapy still seems some way off, progress has been made with precision medicines that target the underlying gene defect and are dependent on the individuals’ particular genotype. The opportunity to correct the CFTR defect in a significant number of patients with CF is now within our reach. However, while these have been met with enthusiasm, we need to ensure that high costs do not prevent access to treatment. At the time of writing, only one example of a precision medicine for CF, ivacaftor, has been funded by the NHS for use in the UK. Further work is required to understand long-term effects of these agents and their impact on the current ‘standard of care’ in the future.
While studies continue to investigate precision medicines, advances continue to be made with drugs intended to ameliorate the symptoms of CF, particularly pulmonary disease. Mucus modifiers and antibiotics will remain the mainstay of treatment for individuals with CF for many years to come. As the number of available antibiotics and mucus modifiers increase, further studies will be required to determine which combination(s) are most appropriate at which point in time, with emphasis on efficacy, tolerability, acceptability and adherence.
CF survival rates in the UK are at an all-time high. With this increased survival, we are seeing an increasing number of individuals with increasingly complex clinical circumstances. In the future, efforts must be focused on supporting adherence to treatments to keep patients with CF well for longer, and make good use of limited healthcare resources. In addition, the affordability of new treatments needs to be taken into account early in the drug development process to protect the viability of conducting studies in the future.
Financial and conflicts of interest disclosure:
The author attended the European CF Society Conference (Brussels, June 2015) at the invitation of Gilead Sciences Ltd (manufacturer of Cayston®) who paid the organisers directly for conference fees, accommodation and travel. The author also attended the UK CF Pharmacist Group Study day (Birmingham, September 2015) — accommodation and travel was covered by Actavis-Allergan (manufacturer of Colomycin®, Colobreathe®, Nebusalâ„¢).
Reading this article counts towards your CPD
You can use the following forms to record your learning and action points from this article from Pharmaceutical Journal Publications.
Your CPD module results are stored against your account here at The Pharmaceutical Journal. You must be registered and logged into the site to do this. To review your module results, go to the ‘My Account’ tab and then ‘My CPD’.
Any training, learning or development activities that you undertake for CPD can also be recorded as evidence as part of your RPS Faculty practice-based portfolio when preparing for Faculty membership. To start your RPS Faculty journey today, access the portfolio and tools at www.rpharms.com/Faculty
If your learning was planned in advance, please click:
If your learning was spontaneous, please click:
References
[1] O’Sullivan BP & Freedman SD. Cystic fibrosis. Lancet 2009;373(9678):1891–1904. doi: 10.1016/S0140-6736(09)60327-5
[2] Cystic Fibrosis Trust. UK Cystic Fibrosis Registry 2015 Annual Data Report. 2016. Available at: https://www.cysticfibrosis.org.uk/~/media/documents/the-work-we-do/uk-cf-registry/2015-registry-annual-data-report.ashx?la=en (accessed August 2016)
[3] Davies JC, Ebdon A & Orchard C. Recent advances in the management of cystic fibrosis. Arch Dis Child 2014;99(11):1033–1036. doi: 10.1136/archdischild-2013-304400
[4] Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration 2000;67(2):117–133. doi: 10.1159/000029497
[5] Brodlie M, Haq IJ, Roberts K et al. Targeted therapies to improve CFTR function in cystic fibrosis. Genome Medicine 2015;7:101. doi: 10.1186/s13073-015-0223-6
[6] Wang Y, Wrennall JA, Cai Z et al. Understanding how cystic fibrosis mutations disrupt CFTR function: from single molecules to animal models. Int J Biochem Cell Biol 2014;52:47–57. doi: 10.1016/j.biocel.2014.04.001
[7] McKone EF, Goss CH & Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest 2006;130(5):1441–1447. doi: 10.1378/chest.130.5.1441
[8] Simmonds NJ, Macneill SJ, Cullinan P et al. Cystic fibrosis and survival to 40 years: a case-control study. Eur Respir J 2010;36(6):1277–1283. doi: 10.1183/09031936.00001710
[9] Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet 2015;16(1):45–56. doi: 10.1038/nrg3849
[10] Cantin AM, Hartl D, Konstan MW et al. Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros 2015;14:419–430. doi: 10.1016/j.jcf.2015.03.003
[11] Stoltz DA, Meyerholz DK & Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med 2015;372:351–362. doi: 10.1056/NEJMc1502191
[12] Vallières E & Elborn JS. Cystic fibrosis gene mutations: evaluation and assessment of disease severity. Adv Genomics Genet 2014;4:161–172. doi: 10.2147/AGG.S53768
[13] Bregnballe V, Schiøtz PO, Boisen KA et al. Barriers to adherence in adolescents and young adults with cystic fibrosis: a questionnaire study in young patients and their parents. Patient Prefer Adherence 2011;5:507–515. doi: 10.2147/PPA.S25308
[14] Cystic Fibrosis Trust. Standards for the Clinical Care of Children and Adults with Cystic Fibrosis in the UK. Second Edition. December 2011. Available at: https://www.cysticfibrosis.org.uk/the-work-we-do/clinical-care/consensus-documents (accessed August 2016)
[15] Döring G, Flume P, Heijerman H et al. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros 2012;11:461–479. doi: 10.1016/j.jcf.2012.10.004
[16] Bhatt JM. Treatment of pulmonary exacerbations in cystic fibrosis. Eur Respir Rev 2013;22:205–216. doi: 10.1183/09059180.00006512
[17] Yang C, Chilvers M, Montgomery M et al. Dornase alfa for cystic fibrosis. Cochrane Database of Systematic Reviews 2016, Issue 4. Art. No.: CD001127. doi: 10.1002/14651858.CD001127.pub3
[18] Dentice R & Elkins M. Timing of dornase alfa inhalation for cystic fibrosis. Cochrane Database of Systematic Reviews 2016, Issue 7. Art. No.: CD007923. doi: 10.1002/14651858.CD007923.pub4
[19] Elkins MR, Robinson M, Rose BRet al.; National Hypertonic Saline in Cystic Fibrosis (NHSCF) Study Group. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006;354(3):229–240. doi: 10.1056/NEJMoa043900
[20] Dentice RL, Elkins MR, Middleton PG et al. A randomised trial of hypertonic saline during hospitalisation for exacerbation of cystic fibrosis. Thorax 2016;71(2):141–147. doi: 10.1136/thoraxjnl-2014-206716
[21] Robinson M, Hemming AL, Regnis JA et al. Effect of increasing doses of hypertonic saline on mucociliary clearance in patients with cystic fibrosis. Thorax 1997;52(10):900–903. doi: 10.1136/thx.52.10.900
[22] Gupta S, Ahmed F, Lodha R et al. Comparison of effects of 3 and 7% hypertonic saline nebulization on lung function in children with cystic fibrosis: a double-blind randomized, controlled trial. J Trop Pediatr 2012;58(5):375–381. doi: 10.1093/tropej/fms004
[23] Bilton D, Robinson P, Cooper P et al.; CF301 Study Investigators. Inhaled dry powder mannitol in cystic fibrosis: an efficacy and safety study. Eur Respir J 2011;38(5):1071–1080. doi: 10.1183/09031936.00187510
[24] Aitken ML, Bellon G, De Boeck Ket al.; CF302 Investigators. Long-term inhaled dry powder mannitol in cystic fibrosis: an international randomized study. Am J Respir Crit Care Med 2012;185(6):645–652. doi: 10.1164/rccm.201109-1666OC
[25] Bilton D, Bellon G, Charlton B et al.; CF301 and CF302 Investigators. Pooled analysis of two large randomised phase III inhaled mannitol studies in cystic fibrosis. J Cyst Fibros 2013;12(4):367–376. doi: 10.1016/j.jcf.2012.11.002
[26] NHS England. Clinical Commissioning Policy: Inhaled Therapy for Adults and Children with Cystic Fibrosis. 2014. Available at: https://www.england.nhs.uk/commissioning/wp-content/uploads/sites/12/2015/01/a01-policy-inhld-thrpy-cf.pdf (accessed August 2016)
[27] Elkins MR, Robinson M, Rose BR et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006;354:229–240. doi: 10.1056/NEJMoa043900
[28] National Institute of Health and Care Excellence (NICE). Mannitol dry powder for inhalation for treating cystic fibrosis NICE technology appraisal guidance (TA266). 2012. Available at: https://www.nice.org.uk/guidance/ta266/chapter/1-Guidance (accessed August 2016)
[29] Langton Hewer SC & Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2014, Issue 11. Art. No.: CD004197. doi: 10.1002/14651858.CD004197.pub4
[30] Tiddens HA, De Boeck K, Clancy JP et al.; ALPINE study investigators. Open label study of inhaled aztreonam for Pseudomonas eradication in children with cystic fibrosis: the ALPINE study. J Cyst Fibros 2015;14(1):111–119.doi: 10.1016/j.jcf.2014.06.003
[31] Pressler T, Bohmova C, Conway S et al. Chronic Pseudomonas aeruginosa infection definition: EuroCareCF Working Group report. J Cyst Fibr 2011;10:S75–S7. doi: 10.1016/S1569-1993(11)60011-8
[32] Littlewood KJ, Higashi K, Jansen JP et al. A network meta-analysis of the efficacy of inhaled antibiotics for chronic Pseudomonas infections in cystic fibrosis. J Cyst Fibros 2012;11(5):419–426. doi: 10.1016/j.jcf.2012.03.010
[33] Schuster A, Haliburn C, Döring G et al; Freedom Study Group. Safety, efficacy and convenience of colistimethate sodium dry powder for inhalation (Colobreathe DPI) in patients with cystic fibrosis: a randomised study. Thorax 2013;68(4):344–350. doi: 10.1136/thoraxjnl-2012-202059
[34] Stuart Elborn J, Geller DE, Conrad D et al. A phase 3, open-label, randomized trial to evaluate the safety and efficacy of levofloxacin inhalation solution (APT-1026) versus tobramycin inhalation solution in stable cystic fibrosis patients. J Cyst Fibros 2015;14(4):507–514. doi: 10.1016/j.jcf.2014.12.013
[35] Deweerdt S. Developing gene therapy to treat cystic fibrosis. The Pharmaceutical Journal 2016;296(7890):340–343.
[36] Davies JC. The future of CFTR modulating therapies for cystic fibrosis. Curr Opin Pulm Med 2015;21:579–584. doi: 10.1097/MCP.0000000000000211
[37] Sermet-Gaudelus I, Boeck KD, Casimir GJ et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med 2010;182(10):1262–1272. doi: 10.1164/rccm.201001-0137OC
[38] National Institute of Health and Care Excellence (NICE). Duchenne muscular dystrophy (nonsense mutation) – ataluren (ID428). 2016. Available at: https://www.nice.org.uk/guidance/indevelopment/gid-duchennemusculardystrophy (accessed August 2016)
[39] Kerem E, Konstan MW, De Boeck K et al. A randomized placebo-controlled trial of ataluren for the treatment of nonsense mutation cystic fibrosis. Lancet Respir Med 2014;2(7):539–547. doi: 10.1016/S2213-2600(14)70100-6
[40] Wilschanski M, Yahav Y, Yaacov Y et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med 2003;349(15):1433–1441.doi: 10.1056/NEJMoa022170
[41] ClinicalTrials.gov. Study of Ataluren in Nonsense Mutation Cystic Fibrosis (ACT CF). Available at: https://clinicaltrials.gov/ct2/show/study/NCT02139306?term=ACT+CF&rank=1&show_locs=Y#locn (accessed August 2016)
[42] Cystic Fibrosis Trust. Ataluren (Translarna) begins assessment journey. 4 May 2016. Available at: https://www.cysticfibrosis.org.uk/news/ataluren-translarna-begins-assessment-journey (accessed July 2016)
[43] Eckford PDW, Li C, Ramjeesingh M et al. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem 2012;287:36639–36649. doi: 10.1074/jbc.M112.393637
[44] Wainwright CE, Elborn JS, Ramsey BW et al.; TRAFFIC Study Group; TRANSPORT Study Group. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for F508del CFTR. N Engl J Med 2016;373(3):220–231. doi: 10.1056/NEJMoa1409547
[45] Vertex Pharmaceuticals Inc. Press Release: Vertex Announces Data from 12-Week Phase 2 Safety Study of VX-661 in Combination with Ivacaftor in People with Cystic Fibrosis Who Have Two Copies of the F508del Mutation. Boston; 2015. Available at: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=902790 (accessed August 2016)
[46] Vertex Pharmaceuticals Inc. Press Release: Vertex Announces Significant Progress in Its Development Efforts to Treat the Cause of Cystic Fibrosis in the Vast Majority of People with the Disease. Phoenix; 2015. Available at: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=935806 (accessed August 2016)
[47] Van Goor F, Hadida S, Grootenhuis PD et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 2009;106(44):18825–18830. doi: 10.1073/pnas.0904709106
[48] Ramsey BW, Davies J, McElvaney NG et al.; VX08-770-102 Study Group. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N Engl J Med 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185
[49] Davies JC, Wainwright CE, Canny GJ et al.; VX08-770-103 (ENVISION) Study Group. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 2013;187(11):1219–1225. doi: 10.1164/rccm.201301-0153OC
[50] De Boeck K, Munck A, Walker S et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros 2014;13(6):674–680. doi: 10.1016/j.jcf.2014.09.005
[51] Davies JC, Cunningham S, Harris WT et al.; KIWI Study Group. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 2016;4(2):107–115. doi: 10.1016/S2213-2600(15)00545-7
[52] McKone EF, Emerson SS, Edwards KL et al. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003;361(9370):1671–1676. doi: 10.1016/S0140-6736(03)13368-5
[53] Moss RB, Flume PA, Elborn JS et al.; VX11-770-110 (KONDUCT) Study Group. Efficacy and safety of ivacaftor treatment: randomized trial in subjects with cystic fibrosis who have an R117H-CFTR mutation. Lancet Respir Med 2015;3(7):524–533. doi: 10.1016/S2213-2600(15)00201-5
[54] Thomspon K, Shaw N, Bentley S et al. Role of the clinical pharmacist in the management of CF. Hospital Pharmacy Europe 2015;80:47–49.
[55] Burgel PR, Bellis G, Olesen HV et al. Future trends in cystic fibrosis demography in 34 European countries. Eur Respir J 2015;46(1):133–141. doi: 10.1183/09031936.00196314
[56] Madge S, Bell SC, Burgel PR et al; ERS/ECFS task force: The provision of care for adults with cystic fibrosis in Europe. Limitations to providing adult cystic fibrosis care in Europe: results of a care centre survey. J Cyst Fibros [In press] 2016. doi: 10.1016/j.jcf.2016.07.001
[57] National Audit Office. Report by the Comptroller and Auditor General: The commissioning of specialised services in the NHS. 2016. Available at: https://www.nao.org.uk/wp-content/uploads/2016/04/The-commissioning-of-specialised-services-in-the-NHS.pdf (accessed August 2016)
[58] Bush A & Simmonds NJ. Hot of the breath: I’ve a cost for’ – the 64 million dollar question. Thorax 2012;67(5):382–384. doi: 10.1136/thoraxjnl-2012-201798
[59] Whiting P, Al M, Burgers L et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost–effectiveness analysis. Health Technol Assess 2014;18(18):1–106. doi: 10.3310/hta18180
[60] NHS England. Clinical Commissioning Policy: Ivacaftor for Cystic Fibrosis (named mutations). 2015. Available at: https://www.england.nhs.uk/commissioning/wp-content/uploads/sites/12/2015/10/a01pc-ivacftr-cystic-fibrosis.pdf (accessed August 2016)
[61] National Institute of Health and Care Excellence (NICE). Lumacaftor–ivacaftor for treating cystic fibrosis homozygous for the F508del mutation NICE technology appraisal guidance (TA398). 2016. Available at: https://www.nice.org.uk/guidance/ta398 (accessed July 2016)
[62] Scottish Medicines Consortium. Lumacaftor 200mg, ivacaftor 125mg film-coated tablet (Orkambi®). SMC No. (1136/16). [online] 2016. Available at: https://www.scottishmedicines.org.uk/files/advice/lumacaftor-ivacaftor__Orkambi__FINAL_April_2016_for_website.pdf (accessed September 2016)
[63] World Medical Association. Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964 and amended by the 64th WMA General Assembly, Fortaleza, Brazil. 2013. Available at: http://www.wma.net/en/30publications/10policies/b3/ (accessed August 2016)
[64] Department of Health. Accelerated Access Review: Interim Report. 2015. Available at: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/471562/AAR_Interim_Report_acc.pdf (accessed August 2016)
[65] European Medicines Agency. Concept paper on the need for revision of the guideline on the clinical development of medicinal products for the treatment of cystic fibrosis (CHMP/EWP/9147/08). 2016. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/08/WC500211478.pdf (accessed September 2016)
[66] National Institute of Health and Care Excellence (NICE). Medicines adherence: involving patients in decisions about prescribed medicines and supporting adherence. NICE guidelines (CG76). 2009. Available at: https://www.nice.org.uk/guidance/cg76 (accessed August 2016)
[67] Quittner AL, Zhang J, Marynchenko M et al. Pulmonary medication adherence and health-care use in cystic fibrosis. Chest 2014;146(1):142–151. doi: 10.1378/chest.13-1926
[68] Eakin MN, Bilderback A, Boyle MP et al. Longitudinal association between medication adherence and lung health in people with cystic fibrosis. J Cyst Fibros 2011;10(4):258–264. doi: 10.1016/j.jcf.2011.03.005
[69] Briesacher BA, Quittner AL, Saiman L et al. Adherence with tobramycin inhaled solution and health care utilization. BMC Pulm Med 2011;11:5. doi: 10.1186/1471-2466-11-5
[70] Mooney K, Ryan C & Downey DG. Pharmacists’ perspectives on monitoring adherence to treatment in cystic fibrosis. Int J Clin Pharm 2016;38(2):296–302. doi: 10.1007/s11096-015-0239-4
[71] Siracusa CM, Ryan J, Burns L et al. Electronic monitoring reveals highly variable adherence patterns in patients prescribed ivacaftor. J Cyst Fibros 2015;14(5):621–626. doi: 10.1016/j.jcf.2015.05.009
[72] Suthoff ED, Bonafede M, Limone B et al. Healthcare resource utilization associated with ivacaftor use in patients with cystic fibrosis. To be published in: J Med Econ. [Preprint] 2016. Available at: http://www.tandfonline.com/doi/full/10.1080/13696998.2016.1178125 (accessed August 2016)
[73] Summary of Product Characteristics (SmPC). Orkambi 200 mg/125 mg film coated tablets (Vertex Pharmaceuticals (Europe) Limited). Updated: 10 August 2016. Available at: www.medicines.org.uk (accessed August 2016)
[74] Edenborough FP, Borgo G, Knoop C et al. Guidelines for the management of pregnancy in women with cystic fibrosis. J Cyst Fibr 2008;7:S2–S32. doi: 10.1016/j.jcf.2007.10.001
[75] Summary of Product Characteristics (SmPC). Pulmozyme 2500 U/ 2.5ml, nebuliser solution Roche Products Limited. Updated: 12 November 2015. Available at: http://www.medicines.org.uk/emc/medicine/1723 (accessed August 2016)
[76] Summary of Product Characteristics (SmPC). Tobi 300 mg/5 ml Nebuliser Solution Novartis Pharmaceuticals UK Ltd. Updated: 16 February 2016. Available at: http://www.medicines.org.uk/emc/medicine/19020 (accessed August 2016)
[77] Summary of Product Characteristics (SmPC). TOBI Podhaler 28 mg inhalation powder, hard capsules Novartis Pharmaceuticals UK Ltd Updated: 16 February 2016. Available at: http://www.medicines.org.uk/emc/medicine/24989 (accessed August 2016)
[78] Summary of Product Characteristics (SmPC). Cayston 75 mg powder and solvent for nebuliser solution (Gilead Sciences Ltd). Updated: 26 May 2016. Available at: http://www.medicines.org.uk/emc/medicine/22358 (accessed August 2016)
[79] Summary of Product Characteristics (SmPC). Colobreathe Forest Laboratories UK Limited (a subsidiary of Actavis PLC) Updated: 12 June 2015. Available at: http://www.medicines.org.uk/emc/medicine/27647 (accessed August 2016)
[80] Summary of Product Characteristics (SmPC). Bronchitol 40 mg inhalation powder, hard capsules Pharmaxis Pharmaceuticals Limited Updated: 29 March 2016. Available at: http://www.medicines.org.uk/emc/medicine/26446 (accessed August 2016)
[81] Summary of Product Characteristics (SmPC). Quinsair 240 mg nebuliser solution. Raptor Pharmaceuticals Europe B.V. Updated: 26 March 2015. Available at: http://www.medicines.org.uk/emc/medicine/32009 (accessed August 2016)
[82] Thompson K. (2016) Approved and off-label drugs for the management of CF in the EU. In: Cystic Fibrosis Pocket Guide. 2nd Ed. Hospital Pharmacy Europe, London. pp49–108.