


Shutterstock.com
Each year sees a couple of dozen new drugs licensed for use, but in their wake there will be tens of thousands of candidate drugs that fell by the wayside. The research and development journey of those new drugs that make it to market will have taken around 12 years and cost around £1.15bn.
The journey will have begun in a university laboratory where researchers, with grants from the research bodies or the pharmaceutical industry, have undertaken basic research to understand the processes behind a disease, often at a cellular or molecular level. It is through better understanding of disease processes and pathways that targets for new treatments are identified. This might be a gene or protein instrumental to the disease process that a new treatment could interfere with, for example, by blocking an essential receptor.
Once a potential target has been identified, researchers will then search for a molecule or compound that acts on this target. Historically, researchers have looked to natural compounds from plants, fungi or marine animals to provide the basis for these candidate drugs but, increasingly, scientists are using knowledge gained from the study of genetics and proteins to create new molecules using computers. As many as 10,000 compounds may be considered and whittled down to just 10 to 20 that could theoretically interfere with the disease process.
The next stage is to confirm that these molecules have an effect and that they are safe. Before any molecules are given to humans, safety and efficacy tests are conducted using computerised models, cells and animals. Around half of candidates make it through this pre-clinical testing stage and these five to 10 remaining compounds are now ready to be tested in humans for the first time. In the UK, approval by the Medicines and Healthcare products Regulatory Agency (MHRA) is required before any testing in humans can occur. The company will put in a clinical trial application (CTA), which will be reviewed by medical and scientific experts, who will decide whether or not sufficient preliminary research has been conducted to allow testing in humans to go ahead.
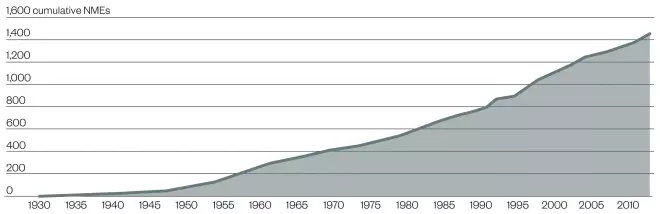
FDA-approved new molecular entities (NME)
Source: FDA / Regulatory Affairs Professional Society
Accumulation of FDA-approved NMEs over time since 1930. The approval of two molecules, morphine and aspirin, pre-dated the creation of the FDA and its precursors
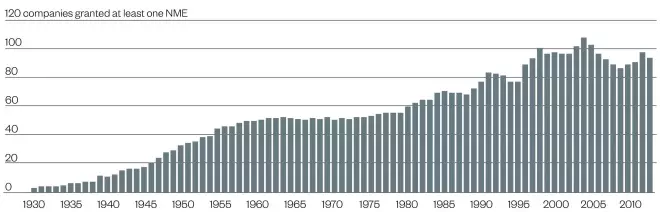
Number of companies with at least one FDA-approved new molecular entity (NME)
Source: FDA / Regulatory Affairs Professional Society
Number of different companies that have gained approval for at least one new molecular entity (NME). As organisations merge or are acquired, the numbers may decrease
Clinical trials
If a CTA application is granted, the safety and pharmacology of a candidate drug will be tested first in a small group of healthy volunteers in a phase 1 trial. Small doses of the compound will be administered to a group of 20 to 100 healthy volunteers who are closely supervised. At least half of compounds will usually be considered safe enough to progress to phase 2 trials.
Phase 2 studies examine the efficacy of a compound in volunteer patients who have the condition the drug is intended to treat. To avoid unnecessarily exposing a volunteer to a potentially harmful substance, these studies use the fewest number of patients possible to provide sufficient statistical power to determine efficacy, usually 100–500 patients, who are monitored and assessed continuously. The aim of phase 2 studies is to determine the most effective dose and method of delivery (for example, oral or intravenous), the appropriate dosing interval, and to reconfirm product safety. Most drugs that fail during clinical trials do so at Phase 2 because they turn out to be ineffective, have safety problems or intolerable side effects.
Those candidates that make it through phase 2 will then be tested in a much larger population of patients in phase 3 trials, often 1,000 to 5,000 across multiple international sites. The aim of these phase 3 trials is to reconfirm the phase 2 findings in a larger population and to identify the best dosage regimen. In doing this the drug company needs to generate sufficient safety and efficacy data to demonstrate an overall risk-benefit for the medicine to allow a submission to be made for a licensing application to the regulatory authority. Despite the rigorous testing that has already taken place, approximately 10% of medicines will still fail at this stage.
Marketing
The process of drug development and marketing authorisation is similar across the world. For those drugs that make it to through phase 3, a submission for marketing authorisations is made to the national regulatory authority in most countries. In the UK, this is the MHRA and, in the US, the Food and Drug Administration (FDA). However, in Europe, drug companies usually now opt to make a central application to the European Medicines Agency (EMA) in order to obtain marketing authorisation for the whole of Europe to avoid having to make multiple applications to individual countries. The submission contains preclinical and clinical information obtained during testing, including information about the chemical makeup and manufacturing process, pharmacology and toxicity of the compound, human pharmacokinetics, results of the clinical trials, and proposed labelling.
If a licence is granted, that is not the end of the process. In England and Wales drug companies need more than a marketing authorisation for most patients to be able to access treatment on the NHS — they also need the National Institute of Health and Care Excellence (NICE) to recommend that it should be made available through the NHS. NICE makes its decisions based on the cost and efficacy of a treatment to determine whether the cost-benefit it offers to the NHS is affordable.
Clinical trials may also continue. Regulatory authorities may insist on phase 4 trials for post-marketing safety surveillance (pharmacovigilance) or they may be undertaken by the company to enable them to target distinct markets. For example, to enable the drug to be used in patients with complex medical problems or pregnant women who are unlikely to have been involved in earlier trials, and to ensure that they do not interact with other drugs.
Patenting
Pharmaceutical companies will patent any molecule that shows promise early in the development process. Patenting prevents other companies copying it for 20 years and covers many aspects of the intellectual property of a drug, including its manufacture, formulation and, in some cases, its use.
The purpose of a patent is to enable the pharmaceutical company that developed it to recoup their development costs and to make a profit to cover the development costs of drugs that failed during the testing process, as well as to invest in the development of future innovative drugs. By the time a drug has undergone the required testing and been licensed, half the patent period will usually have expired.
Once a patent on a drug has expired generic versions of the drug can be manufactured and marketed. For some drugs the period of patent protection can be extended for up to a further five-and-a-half years, so long as this does not take the time in which the drug is under patent protection beyond 15 years after the date it received regulatory approval.
Supporting innovation
As drugs and their development have become more complex and expensive, so have the demands for information from the regulatory agencies. In response, communication channels have opened up between drug companies and regulators well ahead of submissions to help ensure that companies are compiling all the relevant data required for a successful submission. The MHRA has set up a dedicated innovation office to provide advice and support to companies. Its main focus is to aid new drug developers and companies developing unique products such as gene and cell therapy, nanomedicines, or treatments involving new delivery systems or produced through novel manufacturing processes.
Meanwhile NICE offers a fee-based consultancy service to developers of medicines to help them ensure that they generate the evidence they will need to support a NICE evaluation. NICE recommends that any advice is sought after the first human trials to aid planning of the more extensive trial programme.
For every 25,000 compounds that start in the laboratory, 25 are tested in humans, 5 make it to market and just one recoups what was invested. The high cost of current drug development coupled with the trend towards complex medicines and use of genomic markers to predict drug response (personalised medicines) may mean that, in the future, we see a more flexible drug development process and regulatory framework.


