James King-Holmes / Science Photo Library
In 2010, an elderly woman with metastatic bladder cancer who was participating in a clinical trial at Memorial Sloan Kettering Cancer Center (MSKCC) in New York had an extraordinary response to everolimus, a drug that inhibits the mTOR (mammalian target of rapamycin) signalling pathway. The mTOR pathway integrates intracellular and extracellular signals and acts as a central regulator of cell metabolism, growth, proliferation and survival. The woman was the only patient who had complete resolution of metastatic disease out of 37 participants in the trial, and researchers started to look at one gene at a time to try to find out why she responded to treatment. When they found nothing abnormal in the genes they looked at, the team turned to whole-genome sequencing of the tumour to investigate the genetic basis of her remission. They discovered that of the 17,000 mutations located in the genome, one led to a loss-of-function mutation in the tuberous sclerosis complex 1 (TSC1) gene, which correlates with everolimus sensitivity, and was key to her responding to treatment[1]
.
“The reason we could solve that mystery was because we were able to look at every single gene,” says David B Solit, a medical oncologist involved in the trial.
Although whole-genome sequencing is currently too expensive to do in every cancer patient, it is the best way to characterise and identify DNA or RNA sequences so as to discover the acquired alterations that are driving cancer growth. Profiling tumour DNA helps to prioritise anti-cancer treatment by identifying specific therapies to which the patient’s cancer may be sensitive or resistant. Some molecular profiling tests are relatively simple, pointing the proverbial narrow laser beam at selected areas in the genes of interest. But many researchers are now opting to significantly increase the scope of that beam.
“Our approach is to put the flood light on that cancer gene — the entire coding sequence of the gene — to identify additional areas with potential vulnerability to targeted drugs,” says Vincent Miller, a medical oncologist and chief medical officer at Foundation Medicine (Cambridge, Massachussetts), which manufactures two comprehensive genomic-profiling assays.
Gene alterations
Most human tumours are caused by acquired alterations within the cancer genome, leading to oncogene activation or tumour suppressor gene inactivation. Oncogenes are derived from normal genes (proto-oncogenes) involved in the regulation of cell growth. Alterations to the nucleotide sequence, or the amount of protein the proto-oncogene produces, can lead to changes that drive the unchecked growth typical of cancer. Oncogenes can be activated or tumour suppressor genes inactivated in four main ways: single nucleotide polymorphisms, referred to as point mutations, which result from a base substitution at one nucleotide; small duplications of consecutive nucleotides, insertions or deletions involving one or a few nucleotides, or more complex mutations involving a combination of deletions and insertions of one or a few nucleotides; exon or gene copy number changes; and structural variants such as translocations or inversions that often result in fusion genes (see figure).
When Foundation Medicine debuted its FoundationOne assay for malignant solid tumours in 2012, it tested the DNA sequence of 136 genes. Just two years later, the product now tests for alterations in 315 cancer genes, which includes sequencing parts of certain genes, referred to as introns, that aren’t normally coded. The assay — which costs US$5,800 and may not be completely covered by health insurance — reveals all classes of genomic alterations, including base substitutions, insertions, deletions, copy number alterations and selected fusions.
The company reported that more than 9,000 patients were tested with FoundationOne in 2013. In 2014, the projected number is around 25,000. “This is a very brisk ramp for what’s basically a new approach with a steep learning curve,” says Miller. A second assay, FoundationOne Heme, which costs US$7,200 and primarily targets blood cancer and sarcomas, was co-developed with researchers at MSKCC.
Most doctors, explains Miller, only look for a pre-specified, subset of potential mutations, hoping that whichever ones are found inside the tumour’s DNA have been linked to drugs that have demonstrated their effectiveness in fighting those mutations. The downside is that, even when mutations are identified, there is a limited array of treatment. “Metastatic cancer is largely unique among advanced medical conditions, because the therapies are so grossly inadequate that most treatments — even the good ones — fail,” says Miller.
This cancer-drug-finding challenge has somehow turned the task of looking for the oddball mutation in specific types of cancer genes — alterations that would not normally be found in that tumour type — into a promising cancer-fighting strategy.
When an assay uses an all-inclusive approach and ignores a mutation’s proclivity for certain types of cancer, a mutation — with a good-outcome drug associated with it — may turn up in the most unexpected tumours.
As an example, Miller cites Herceptin (trastuzumab), a targeted therapy for an amplification of the human epidermal growth factor receptor 2 (HER2) gene often found in HER2-positive metastatic breast cancer and HER2-positive gastric cancer. “We have now seen — because we conduct the same broad test in every tumour type — either an amplification or mutation in this critical HER2 gene in nearly 30 different tumour types,” he says.
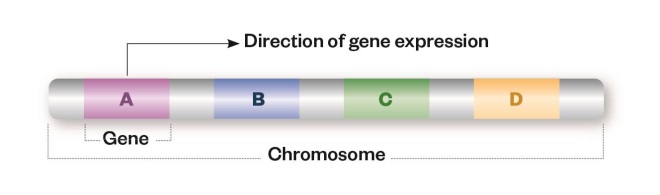
Genomic alterations
Cancer is caused by alterations to the genes that regulate cell growth and division. These alterations can affect the nucleotide sequence of the gene or the structure of the whole chromosome.
Reference
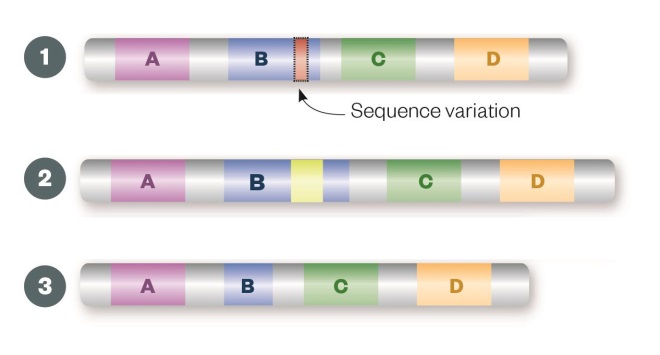
Sequence variation
Single nucleotide polymorphisms (1), insertions (2) or deletions (3) may result in a change to the amino acid sequence of the encoded protein (missense mutation) or premature truncation (nonsense mutation), which can render the resulting protein non-functional.

Structural variation
Large insertions (1), deletions (2) or duplications (3) of the entire gene can result in gene copy number changes leading to alterations in the levels of gene expression and overproduction or underproduction of the resulting protein. Inversions (4) or translocations (5) of a segment of the chromosome can create fusion genes, which may lead to a protein with a different function from the two fusion partners, or to up-regulation of the protein.
The technology
Although the logic behind these mutation search missions seems simple, the technology is not. The DNA of a malignant tumour is more than just cancerous cells. It’s a mixture of cancer cells — sometimes as little as 20% — and the normal tissue of that organ, including white blood cells, collagen, fibroblasts and many other components.
“The critical point to make is that cancers are impure and they also contain a lot of non-cancerous cells,” says Miller. “That’s why it’s imperative not only to have an incredibly sensitive, specific and robust assay, but also to have very sophisticated computational algorithms, which allow computational biologists to determine if what they’re seeing is likely a true mutation or a normal variant.”
If either the assay or the algorithms are wanting, he says, then you have inconsistencies among clinical laboratories, which are very difficult to reconcile. “That’s why we spend a lot of time and money developing and publishing rigorous, analytic validations around our tests,” says Miller.
Solit, who is director of the Center of Molecular Oncology at MSKCC, also considers time and money to be important factors in the field of cancer genome sequencing. “We have the technology to figure out the mutations in every patient’s cancer, but what’s holding us up now is the ability to do these tests on everyone because it’s either too expensive or time-consuming,” he says.
As recently as five years ago, these tests could only be done one gene at a time. But next-generation sequencing methods allow for hundreds of genes to be looked at simultaneously, more quickly and at a dramatically lower cost. As the technology gets better, the hope, says Solit, is that we’ll soon be able to solve every cancer case. The key, he says, is figuring out what the important mutations are in a particular tumour, which often means taking that floodlight approach and sequencing the entire genome “in one sitting”.
“One can envision that in 10 or 15 years, or sooner, it’s going to cost less than US$1,000 to sequence the entire genome of every patient’s tumour.”
But whole-genome sequencing of tumours does not come cheap. In 2011, the technical cost, without the analysis, was around US$20,000. “Now it’s down to about US$4,000 per whole genome,” says Solit. “It’s still too expensive to do in every patient but it’s getting less expensive every year. One can envision that in 10 or 15 years, or sooner, it’s going to cost less than US$1,000 to sequence the entire genome of every patient’s tumour.”
Now, at MSKCC, patients with advanced metastatic cancer who have had their tumours genetically tested fall into one of three groups, says Solit. In the first group are patients whose cancer has a mutation for which there is an associated drug, which is prescribed for the patient. In the second group are patients whose cancer is found to have an activating driver mutation for which there is no associated drug. “Patients in this group may wind up in clinical trials that MSKCC may be offering,” he says. “Even though the patient is disappointed there’s no standard, approved drug to target the specific mutation, at least physicians can avoid treating them with a drug that has no chance of working, based on the assay.” Finally, in the third group are patients whose tumours have no mutations of any of the 341 most commonly mutated cancer-associated genes.
As more tumours are sequenced, Solit anticipates that the number of patients in the third group will shrink dramatically because the research will have advanced to find key mutations in all patients. And, with the development, regulation and approval of additional cancer drugs, the number of patients in the second group will also decrease.
But if resourcing time and money is a challenge, so is the development of effective therapies. Solit points to the massive, international, and so far unsuccessful effort over the past 30 years to create a KRAS inhibitor. “Between 15% and 20% of patients have KRAS gene mutations, which are common in various cancers, and we still don’t have a good KRAS inhibitor,” he says. “There are a lot of cancer genes which are important, and we don’t have drugs to target them, either.”
Biological factors
For some experts, wiping out cancer goes beyond sequencing and digs into the cunning core of cancer itself.
“Cancer tumour sequencing gives us information about how the DNA has been altered, but that’s just one part of the picture,” says Ilya Shmulevich, a researcher specialising in computational biology at the Institute for Systems Biology, a non-profit research institution in Seattle, Washington. “Ultimately, what’s needed is prediction, and to do that we must model a tumour on multiple scales of biological organisation. This involves not only DNA but also measurements found through RNA sequencing, DNA methylation and protein expression.”
Shmulevich says that applying these biological factors in the tumour model allows researchers to paint a more complete view of the tumour environment and to better simulate how the entire tumour behaves. This, in turn, provides a way for scientists to anticipate whether use of a specific drug might have a beneficial outcome. “What goes on inside a tumour is a terribly complex problem and understanding oncogenesis — by profiling tumours using multiple technologies — is just as important as developing new drugs.”
Different institutions may be taking different technical approaches to genetic analysis of human tumours, but all their efforts are towards one end. “The common and final goal is to identify the driver or key mutation in each patient,” says Solit. “Every major cancer centre is trying to put into place a programme to genetically test tumours. This is the future, and eventually it will become the standard for each patient.”
Coeli Carr is a freelance writer based in New York.
References
[1] Iyer G, Hanrahan AJ, Milowsky MI et al. Genome sequencing identifies a basis for everolimus sensitivity. Science 2012;338:221.