Summary
Adult myeloid leukaemias and the myelodysplastic syndromes (MDS) are all disorders of bone marrow stem cells that result in ineffective haematopoiesis. Patients with acute myeloid leukaemia usually present with symptoms associated with bone marrow failure; MDS and chronic myeloid leukaemia are often detected by chance.
Diagnosis generally relies on a mixture of microscopy and cytogenetic analyses. An increasing understanding of the genetic abnormalities associated with these diseases has led to improvements in risk stratification and treatment.
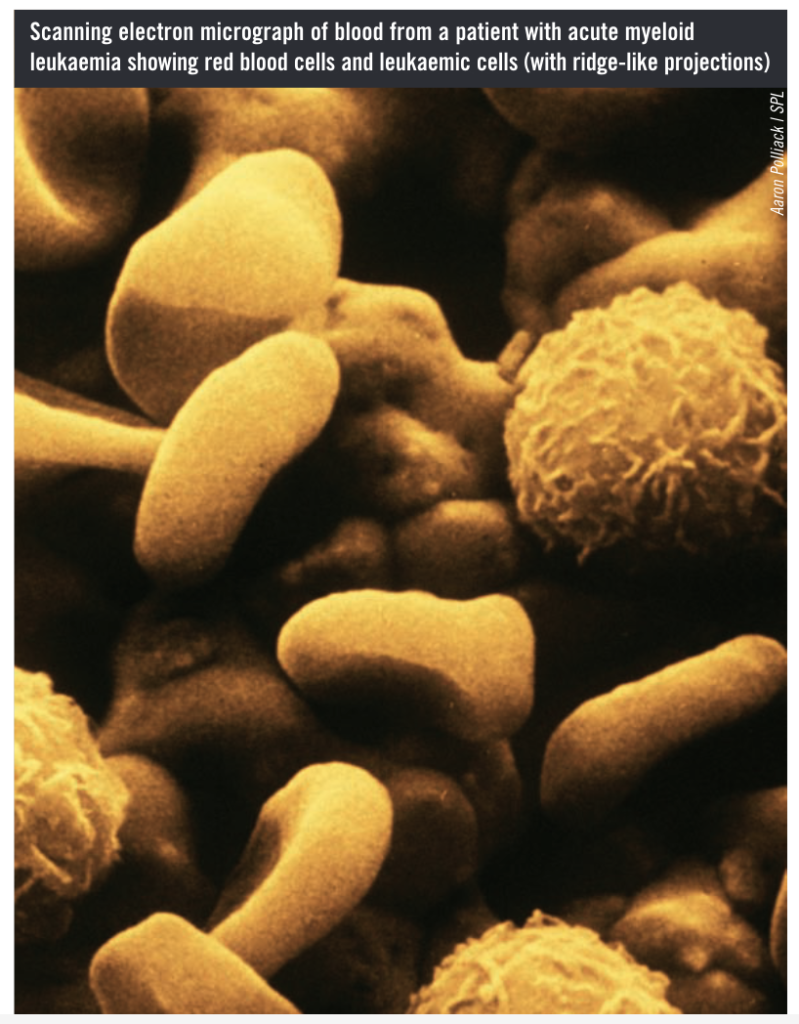
Leukaemia is a cancer of the bone marrow and blood characterised by abnormal differentiation of haematopoietic stem cells and subsequent over proliferation of blood cells that cannot properly mature. Myeloid leukaemias (also termed myelogenous or granulocytic leukaemias) usually involve the over production of immature granulocytic white blood cells, known as blasts (in contrast to lymphoblastic leukaemias, which affect lymphocyte proliferation). The immature granulocytes do not function like mature, healthy white blood cells; they also overcrowd the bone marrow, preventing normal production of red blood cells and platelets. (Some rarer forms of myeloid leukaemia involve the over proliferation of immature red blood cells or platelets.)
This article focuses on acute myeloid leukaemia(AML) and chronic myeloid leukaemia (CML), as well as the myelodysplastic syndromes (referred to as MDS). Although MDS is not classified as a leukaemia, it may be considered to be a pre-leukaemic condition. MDS is a bone marrow stem cell disorder that results in ineffective or disordered myeloid cell formation; it causes progressive bone marrow failure and leads to AML in 30% of cases.
During recent years, significant progress has been made in understanding the development of these diseases at a molecular level; this has led to important advances in risk stratification and treatment strategies. Treatment is discussed in an accompanying article.
Epidemiology and causes
AML is the most common type of acute leukaemia diagnosed in adults, with five to eight new cases being reported in Europe each year per 100,000 population.1
The incidence increases rapidly with age and is closer to 20new cases per year per 100,000 population among those aged over 70 years.2 MDS has an annual incidence of four cases per 100,000 population,3 but many cases are believed to be undiagnosed, particularly among the elderly, so the true incidence is likely to be higher. CML is less common — diagnosed in one to two adults per year per 100,000 population, with a median age of onset of about 60 years.4
For most cases of AML, CML and MDS the cause is unknown, but certain risk factors (eg, exposure to ionising radiation) have been identified. Furthermore, previous exposure to chemotherapy (particularly alkylating agents and topoisomerase-II inhibitors) can predispose individuals to secondary MDS and AML. A link has also been demonstrated between exposure to benzene, pesticides, herbicides and some dyes and the development of AML. Certain genetic disorders (eg, Down’s syndrome, Fanconi anaemia) are also associated with an increased risk of AML.
Clinical presentation
Patients with AML typically present with fatigue, dyspnoea, bruising or bleeding, or signs of infection, all of which are direct consequences of bone marrow failure.Bleeding due to disseminated intravascular coagulation(the widespread formation of clots in the bloodstream that consumes platelet stores and leads to uncontrolled bleeding) is often seen in patients presenting with acute promyelocytic leukaemia (a form of AML).
The abnormal blood cell differentiation and high rates of apoptosis typical of MDS mean that patients usually present with one or more peripheral blood cytopenias. Consequently, although many patients with MDS do not appear unwell when diagnosed, symptoms related to anaemia, neutropenia or thrombocytopenia may be apparent.
CML can be separated into three distinct phases, which are set out in Box 1. Most patients with CML present during the chronic phase of the disease. Around50% of these patients are asymptomatic and the disease is diagnosed by chance following a routine blood test for another condition. Patients who are symptomatic may present with one or more of the following:5
- Malaise and fatigue
- Weight loss
- Unexpected bruising and bleeding
- Abdominal discomfort (often due to enlarged spleen)
- Bone tenderness
- Excessive sweating
- Hepatomegaly
- Purpura
In the advanced phases of CML, patients may present with fever, cachexia (severe weight loss) and an enlarged spleen.

Diagnosis
Whenever AML, CML or MDS is suspected, a bone marrow aspirate and trephine biopsy (which removes a small amount of solid marrow as well as bone marrow cells) should be obtained for analysis. In MDS, abnormally differentiated cells will be present in the bone marrow and the blast percentage will be below 20%.
A blast percentage above 20% indicates a diagnosis of acute leukaemia.6 Immunophenotypic and cytochemic alanalyses are carried out to determine whether the leukaemia is myeloid or lymphoblastic — for example, a positive blast stain using myeloperoxidase or Sudan BlackB indicates AML. Further cytogenetic and molecular analyses are used to classify patients into prognostic risk groups (see below).
Patients presenting with CML usually have an excess of immature granulocytes visible on microscopy. Blood tests often show low haemoglobin (9–12g/dl) and a markedly raised white cell count (25–1,000 x109/L; normal = 4–11 x109/L). Platelet counts can vary — 10%of patients have a count lower than 150 x109/L, 50% of patients have a count above 400 x109/L, while the rest lie somewhere in between.
Genetic abnormalities
Chromosomal analysis to identify genetic abnormalities is important for diagnosis and estimating prognosis of patients diagnosed with AML, CML or MDS.
AML
In the case of AML, the most common mutation occurs in the FLT3 tyrosine kinase receptor gene.7 This mutation is associated with a poor prognosis and is beingtargeted specifically by a number of novel therapies for AML.
A translocation of genetic material between chromosomes 15 and 17 (t[15:17]) is characteristic of acute promyelocytic leukaemia and results in disrupted cell differentiation via a mutation in the retinoic acid receptor.
MDS
The pathogenesis of MDS is not well understood but several genetic mutations have been implicated in causing abnormalities in haematopoiesis. Chromosomal abnormalities are identified in 40–70% of patients with primary MDS.8 Although the presence of multiple chromosomal abnormalities is associated with poorer outcomes among patients with MDS, some abnormalities confer a survival advantage. For example, patients with a deletion in the long arm of chromosome 5 (del 5q) have a better prognosis than patients with a normal karyotype.8 Del 5q affects 20–30% of those with MDS. In some people the number of mutations increases over time and the risk of progression to AML increases accordingly.
CML
Almost all patients with CML carry a chromosomal abnormality known as the Philadelphia chromosome. It is the result of a translocation of DNA between chromosomes 9 and 22 (t[9:22]), leading to a lengthened chromosome 9 and a shortened chromosome 22 (the latter is the Philadelphia chromosome).9The deregulated proliferative activity of BCR-ABL tyrosine kinase, which is expressed by a hybrid gene (BCR-ABL) on thePhiladelphia chromosome, is central to the development of chronic phase CML. Chromosomal analysis to check for the presence of the Philadelphia chromosome is part of the diagnostic process for suspected CML.It is believed that further genetic mutations lead to progression into the accelerated and blastic phases of CML.
Disease classification and risk grouping
AML
Historically, AML has been classified according to the French-American-British (FAB) criteria, which divided sufferers of the disease into six main subgroups (AML M1–6).
However, from a prognostic point of view, the most important predictors of outcome for patients with AML are described in Box 2. Based on cytogenetics alone, three prognostic groups (good risk, intermediate risk and poor risk) can be defined and these allow treatment to be tailored appropriately.11For example, a patient with “good risk” cytogenetic results would be given chemotherapy treatment alone because the survival rate for this risk group with such treatment is 60–70%. However, a patient with a “poor risk” cytogenetic profile has a predicted long-term survival of less than 20% with chemotherapy alone, so would be considered for an allogeneic stem cell transplant if he or she achieved a complete response with induction chemotherapy.
MDS
MDS can be subclassified according to two international classification systems —a FAB classification12 and a more recent World Health Organization classification.6
Although these classifications have some prognostic value, a more accurate predictor of outcome is the international prognostic scoring system (IPSS), based on karyotype, level of blasts in the bone marrow and the presence of cytopenias.13 By allocating a score to each ofthese parameters, patients can be classified into one offour prognostic groups:
- Low risk
- Intermediate-1 risk (Int-1)
- Intermediate-2 risk (Int-2)
- High risk
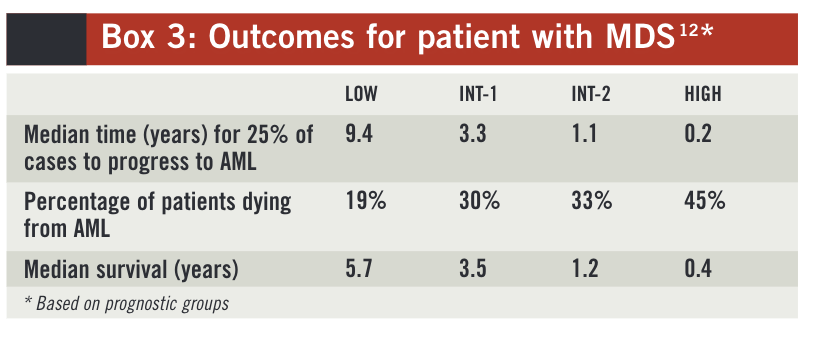
Outcome predictions based on these prognostic groups are set out in Box 3.
CML
As described in Box 1, CML is divided into chronic, accelerated and blastic phases. Scoring systems such as theSokol score14have been used to estimate prognosis for patients with CML based on their age, spleen size, blast percentage and platelet count. However, the usefulness of such scoring systems as prognostic tests could diminish now that highly effective first-line treatments for CML are available. Such treatments are discussed in the accompanying article

References
1 Fey M, Dreyling M. Acute myeloblastic leukemia in adult patients: ESMO clinical recommendations for diagnosis, treatment and follow-up. Annal sof Oncology 2009;20 (s4):100–1.
2 Craddock C. Acute leukaemias. Medicine 2009;37:190–4.
3 Bowen D, Culligan D, Jowitt S, et al. Guidelines for the diagnosis andtreatment of adult myelodysplastic syndromes. British Journal of Haematology 2003;120:187–200.
4 Baccarani M, Dreyling M. Chronic myelogenous leukemia: ESMO clinical recommendations for diagnosis, treatment and follow-up. Annals ofOncology 2009;20 (s4):105–7.
5 Leukaemia. In: Tobias J, Hochhauser D. Cancer and its management. 6th edition. Oxford: Wiley Blackwell; 2010.
6 Vardiman J, Harris N, Bruning R. The World Health Organization classification of myeloid neoplasms. Blood 2002;100:2292–302.
7 Kottaridis P, Gale R, Frew M, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukaemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from theUnited Kingdom Medical Research Council AML 10 and 12 trials. Blood 2001;98:1752–9.
8 Nimer S. Clinical management of myelodysplastic syndromes with interstitial deletion of chromosome 5q. Journal of Clinical Oncology 2006;24:2576–82.
9 Goldman J. Chronic myeloid leukaemia — advances in biology and new approaches to treatment. New England Journal of Medicine 2003;349:1451–64.
10 Acute Leukaemias. In: Hoffbrand A, Moss P, Pettit J. Essential Haematology. 5th edition. Oxford: Wiley Blackwell; 2006.
11 Grimwade D, Hills R. Independent prognostic factors for AML outcome.Haematology ASH Educational Program 2009:385–93.
12 Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. British Journal of Haematology1982;51:189–99.
13 Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood1997;89:2079–88.
14 Sokal J, Cox E, Baccarani M, et al. Prognostic discrimination in ‘goodrisk’ chronic granulocytic leukemia. Blood 1984;63:789–99.