Maurizio de Angelis / Science Photo Library
In 2014, the National Institute for Health and Care Excellence (NICE), England’s health technology assessment body, stated in its guidance on cardiovascular risk assessment and modification of blood lipids for the primary and secondary prevention of cardiovascular disease (CVD) that “fibrates should not be routinely offered, alone, or in combination with statins, for the primary or secondary prevention of CVD”[1]
. The guidance also stated that “nicotinic acid, bile acid sequestrants and omega-3 fatty acids should not be offered, alone, or in combination with statins, for the primary or secondary prevention of CVD”[1]
. Ezetimibe retains a role in line with the recommendations in NICE TA385[2]
. However, this has led healthcare practitioners to question the role of the pharmacological alternatives to statins in clinical practice. This article will discuss the place of non-statin drugs in the management of dyslipidaemia in adults, which should be largely performed under the specialist supervision of, and in consultation with, a lipidologist.
Dyslipidaemia refers to a spectrum of lipid disorders, including excessive levels of certain lipoproteins, such as low density lipoprotein cholesterol (LDL-C), low levels of other lipoproteins, such as high density lipoprotein cholesterol (HDL-C), abnormalities in the composition of the various lipoprotein particles or high levels of triglycerides. More information, including the types and relative size of the four major lipoprotein classes, can be found in ‘Table 1: Major lipoprotein classes’. Primary dyslipidaemias include those that are caused by an underlying genetic defect (see ‘Box 1: Primary disorders of lipid metabolism’), while secondary dyslipidaemias occur as a result of identifiable and reversible causes, including hypothyroidism, chronic kidney disease, diabetes, nephrotic syndrome, obesity, excess alcohol intake or drug therapies[3]
.
| Table 1: Major lipoprotein classes Non-HDL cholesterol = Total Cholesterol – HDL cholesterol | ||||
|---|---|---|---|---|
| Information from: Jairam V, Uchida K & Narayanaswami V. Lipoproteins — Role in Health and Diseases. Chapter 16: Pathophysiology of Lipoprotein Oxidation; and German JB, Smilowitz JT & Zivkovica AM. Curr Opin Colloid Interface Sci. 2006;11:171–183. | ||||
| High density lipoprotein | Low density lipoprotein | Very low density lipoprotein | Chylomicrons | |
| Major core lipids | Cholesterylester | Cholesterylester | Triglyderide | Triglyderide |
| Relative size (approx) | 7–13nm | 21–27nm | 30–90nm | 200–600nm |
| Role | Anti-atherogenic | Pro-atherogenic | Pro-atherogenic | Pro-atherogenic |
It is well established that elevated concentrations of total cholesterol and LDL-C increase the risk of coronary heart disease (CHD), while HDL-C confers protection. Understanding and managing dyslipidaemia appropriately is therefore of particular importance to reduce cardiovascular events in patients who have, or are at risk of, CVD, but dyslipidaemia can also predispose patients to other disorders, for example, severe hypertriglyceridemia (>10mmol/L) is responsible for up to 4% of cases of acute pancreatitis[4]
.
Primary familial hyperlipidaemias, specifically familial combined hyperlipidaemia (FCH) and familial hypercholesterolaemia (FH), are inherited conditions where the underlying defect relates to a disorder of lipid metabolism (see ‘Box 1: Primary disorders of lipid metabolism’). The frequency of these conditions in the general population varies from 1 in 500 to 1 in a million depending on the underlying defect in lipid metabolism[5]
. A Danish study indicates a possible higher prevalence of 1 in 270[6]
. NICE estimates that 106,000 people in England are affected by FH. Dyslipidaemia, including the types and pathophysiology, has been the focus of a learning article found here
[7]
.
Box 1: Primary disorders of lipid metabolism
- Familial hypercholesterolaemia (FH): Excess circulating low density lipoprotein cholesterol (LDL-C) and total cholesterol from conception, resulting in premature cardiovascular disease (CVD). In the more common heterozygous FH, there is a gene defect on only one of the chromosomes[5]
. The National Institute for Health and Care Excellence estimates heterozygous FH affects 1 in 500 people in the UK (approximately 110,000 individuals)[5]
. However, more recent data suggest a higher prevalence, with a Danish study of more than 69,000 people detecting a prevalence of 1 in 137[6]
. There are many potential gene mutations but they most commonly affect the LDL-C receptor, resulting in cholesterol levels two or three times higher than average for the general population. Homozygous FH is a very rare metabolic disorder, affecting 1 per million of the population, where the individual inherits affected gene mutations on both chromosomes, resulting in an inability to clear LDL-C particles from the circulation. This leads to very high cholesterol levels and results in CVD being expressed at a very young age — myocardial infarction has been reported in children aged as young as two or three years — and a much reduced lifespan if untreated, with fatal cardiovascular events commonly observed when patients are in their late teens and early 20s[8]
. - Familial combined hyperlipidaemia: Results in excessive synthesis VLDL-C and predisposes the individual to premature CVD[3]
- Familial type III hyperlipoproteinaemia: Results in accumulation of chylomicron and VLDL remnants and predisposes the individual to premature CVD[3]
- Familial lipoprotein lipase deficiency: Results in elevated triglycerides and chylomicron concentrations associated with acute pancreatitis[3]
- Familial apolipoprotein C-II deficiency: Results in lack of lipoprotein lipase and a resulting increase in triglyceride, predisposing the individual to acute pancreatitis. Premature atherosclerosis is unusual but has been described[3]
NICE guidance recommends that an underlying familial hyperlipidaemia is considered in any individual with a total cholesterol >7.5mmol/L with evidence of premature CVD present in the family (e.g. myocardial infarction or stroke in a first degree relative aged under 60 years), as well as in those with a baseline total cholesterol >9mmol/L or a non‑HDL cholesterol concentration of more than 7.5mmol/L, even in the absence of a family history of premature CVD[1],[5]
. Healthcare professionals should exclude secondary causes of hypercholesterolaemia before a diagnosis of FH is considered. The diagnosis of FH should be confirmed using the Simon Broome criteria, which includes a combination of the family history, any clinical signs (specifically tendon xanthomata), cholesterol concentration and, where available, DNA testing[5]
.
As dyslipidaemias are mainly asymptomatic until the first significant cardiovascular event occurs, many people with FH in the UK are currently undetected; with only 15% of the predicted FH population currently identified[9]
. Detection of hyperlipidaemia, for example through the NHS Health Check, is desirable as it allows initiation of drug therapy and even interventional procedures to manage lipids and prevent development of atherosclerotic disease. Once an index case of FH has been identified, cascade screening should take place to identify any other family members with the condition and facilitate early treatment[5]
.
A definition of non-familial hypercholesterolemia is elusive in the literature, but could be defined as the presence of raised lipid levels in the absence of any premature CVD (aged <60 years) within the family. The total cholesterol threshold for a diagnosis of non-familial hypercholesterolemia remains a matter for debate, with some advocating a cut off total cholesterol >7mmol/L and others advocating total cholesterol >6.5mmol/L or 5.5mmol/L[10]
. NICE estimates that primary non-familial hypercholesterolaemia affects an estimated 1.5 million people in England.
Treatment of dyslipidaemias
Statin therapy remains the first-line treatment for patients with the vast majority of dyslipidaemias. NICE FH guidance recommends that healthcare professionals should consider prescribing a high-intensity statin at the maximum licensed dose (e.g. atorvastatin 80mg daily) or at the maximum tolerated dose to achieve a reduction in LDL-C concentration of greater than 50% from baseline[5]
. On account of the high levels of cholesterol present in many dyslipidaemias, statins alone — even high intensity agents at the maximum doses — are insufficient to reduce cholesterol to acceptable levels. In addition, many patients have contradictions to statins, cannot tolerate statins, or have intolerance that limits the statin dose. It is in these specific cohorts of patients that other agents may be required.
Ezetimibe
When ezetimibe is used, it blocks cholesterol reabsorption from the gastrointestinal tract by acting on the cholesterol transporter located within with the intestinal brush border. Ezetimibe monotherapy reduces LDL-C by 15–20%, with a small increase in HDL-C and a reduction in triglycerides. Ezetimibe monotherapy is recommended by NICE for the treatment of primary hypercholesterolaemia in patients where initial statin therapy is contraindicated or not tolerated[2]
. Ezetimibe plus statin therapy is recommended for the treatment of primary hypercholesterolaemia where the total cholesterol or LDL-cholesterol is not controlled, either after dose optimisation of statin therapy or where the dose is limited by intolerance and a change to an alternative statin is being considered[2]
.
Ezetimibe in combination with statin therapy is recommended in patients whose serum total or LDL-C concentration is not appropriately controlled (either after appropriate dose titration of initial statin therapy or because dose titration is limited by intolerance to the initial statin therapy)[2]
.
When added to a statin, ezetimibe lowers LDL-C by approximately 20% more than is achieved with statin therapy alone[11]
.
Ezetimibe use has been limited because of a lack of cardiovascular outcomes data. Recent publication of the IMPROVE-IT study may have clinical implications and could change perceptions of the drug. Over a period of seven years, the addition of ezetimibe to simvastatin 40mg in 18,144 patients following an acute coronary syndrome reduced the primary end point — a composite of cardiovascular death, myocardial infarction, unstable angina requiring rehospitalisation, coronary revascularisation, or stroke — by 6.4% when compared with patients who received simvastatin alone (P =0.016). The absolute reduction in risk over seven years was 2.0%, with 32.7% in the ezetimibe/simvastatin arm experiencing a primary end point compared with 34.7% in the simvastatin arm[12]
.
Ezetimibe should be prescribed at a dose of 10mg daily and can be taken at any time of the day, with or without food. Ezetimibe is contraindicated in patients who have a hypersensitivity to the active substance or excipients, are pregnant or lactating, and, if co-administered with a statin, in patients with active liver disease or unexplained persistent elevations in serum transaminases. When used alone or in combination with a statin, it is generally well tolerated with a side effect profile similar to placebo[13]
.
Fibrates
The use of fibrates has waned over the past 10–15 years because of lack of efficacy data compared with statins, but some agents from this class, such as bezafibrate and fenofibrate, are still used in selected patients. Fibrates modify the expressions of genes involved in lipoprotein metabolism and primarily lower triglycerides, with a lesser reduction in LDL-C and small increases in HDL-C. Early data for fibrate monotherapy in primary and secondary prevention studies showed promise with a significant reduction in coronary events and stroke and even a modest impact on cardiovascular mortality[14],[15]
. However, the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study compared fenofibrate to placebo in a diabetic population on baseline statin therapy but failed to demonstrate a benefit from the addition of fibrate on the primary end point of coronary events, including coronary heart disease death or non-fatal myocardial infarction. However, it did show a small but significant reduction of 21% in the secondary end point of cardiovascular events, including death, myocardial infarction, stroke, and coronary or cardiovascular revascularisation[16]
.
Similarly, the ACCORD study compared combination statins plus fibrate therapy to statins alone in patients with type 2 diabetes and concluded that the combination of fenofibrate and simvastatin did not reduce the rate of fatal cardiovascular events, non-fatal myocardial infarction, or non-fatal stroke, compared with simvastatin alone. These results do not support the routine use of combination therapy with fenofibrate and simvastatin to reduce cardiovascular risk in the majority of high-risk patients with type 2 diabetes[17]
.
While NICE guidance recommends that fibrates are not prescribed routinely in the primary and secondary prevention of CVD, fibrates may retain a role in patients with dyslipidaemias in specialist lipid clinics, particularly in patients with high levels of triglycerides (>10mmol/L)[1]
. While the results of the FIELD study were disappointing overall, they did confirm the safety of using fenofibrate in combination with a statin, although there is still a risk of adverse effects, including rhabomyolysis, when prescribing the combination[16]
. Gemfibrozil should not be used with a statin because of the higher risk of myopathy[18]
. Fibrates may also be considered for first-line use in patients with isolated hypertriglyceridaemia and in individuals with mixed dyslipidaemia when a statin is contraindicated or not tolerated[19],[20]
. For examples of fibrates prescribed in clinical practice and their usual maintenance doses, see ‘Table 2: Fibrates prescribed in clinical practice’.
| Table 2: Fibrates prescribed in clinical practice | |
|---|---|
| Usual maintenance dose | |
| Benzafibrate[20] | 400–600mg daily (depending on formulation) |
| Fenofibrate[19] | 200–267mg daily |
Fibrates are contraindicated in significant hepatic disease (other than fatty infiltration of the liver associated with raised triglyceride levels), gallbladder diseases with or without cholelithiasis, nephrotic syndrome, renal failure with serum creatinine >135µmol/L or creatinine clearance <60ml/min, patients undergoing dialysis, patients with known hypersensitivity to fibrate or excipients, and patients with known photoallergic or phototoxic reactions to fibrates. Gastrointestinal symptoms, such as nausea, diarrhoea and abdominal pain, are common during fibrate therapy, particularly on initiation. Myositis has been described and is associated with muscle pain, unusual tiredness or weakness[19],[20]
.
Bile acid binders
This drug class acts by binding bile acids in the intestine and thereby preventing reabsorption. This stimulates the conversion of cholesterol to bile acids and encourages the removal of circulating LDL-C. Bile acid binding agents reduce total cholesterol and LDL-C, but can increase triglyceride levels.
Cholestyramine and cholestipol demonstrated a trend towards reduced mortality and non-fatal myocardial infarction in one trial[21]
but their use is limited in clinical practice by poor tolerability, drug interactions and increases in circulating triglyceride levels. For examples of bile acid binders and their usual maintenance doses, see ‘Table 3: Bile acid binders prescribed in clinical practice’.
| Table 3: Bile acid binders prescribed in clinical practice | |
|---|---|
| Usual maintenance doses | |
| Cholestyramine[22] | 3–6 sachets daily |
| Colestipol[23] | 5–30g daily in one or two divided doses |
| Colesevalam 625mg tablets[24] | 4–6 tablets daily |
Bile acid binders are contraindicated if there is known hypersensitivity to the active substance or to any excipients listed and where there is bowel or biliary obstruction[23]
.
Gastrointestinal side effects, such as bloating, flatulence, heartburn and constipation, are commonly reported. Long-term administration can interfere with the absorption of fat soluble vitamins (A, D and K) and folic acid, and supplementation may be required[23]
. Drug interactions are common so other medicines should be taken at least one hour before or four hours after the bile acid binding agent[23],[24],[25]
.
Colesevelam, launched in 2007, shows some efficacy and improved tolerability but the possibility of drug interactions remains, and a negative impact on triglyceride levels has already been established[24]
. This class should only be considered as an adjunctive therapy under specialist lipidology advice where first-line options for the management of dyslipidaemias are contraindicated or ineffective[5]
.
Nicotinic acid derivatives
While nicotinic derivative did look promising in terms of effects on the lipid profile (e.g. reduced total cholesterol, LDL-C, triglycerides, increased HDL-C), these products have recently been withdrawn from the market in the UK on account of safety concerns following a meta-analysis that indicated an increased risk of diabetes, myositis and infection[26]
. Only patients under the management of a lipid specialist should be prescribed nicotinic acid derivatives, in line with NICE guidance for FH, which must be imported on a named patient basis[5]
.
High doses of nicotinic acid (1.5–2g daily) are required to achieve a 15% reduction in LDL-C levels[9]
, with doses of up to 6g daily given to manage severe hypertriglyceridaemia[13]
. However, patient adherence to these doses is often poor because of prostaglandin mediated side effects, such as flushing[25]
. Aspirin or ibuprofen can be coadministered to attenuate the flushing[25]
. Extended release nicotinic acid was recently investigated in the HPS2-THRIVE study, in which the drug was combined with laropiprant (an anti-flushing agent). The addition of extended-release nicotinic acid–laropiprant to statin-based LDL cholesterol–lowering therapy in HPS-THRIVE did not significantly reduce the risk of major vascular events but increased the risk of serious adverse events[27]
. As a result, the European Medicines Agency (EMA), which evaluates medicinal products for use in Europe, suspended the license for Tredaptive (nicotinic acid/laropiprant) across Europe[28]
.
Omega-3 fatty acid supplements
Primarily used to manage patients with hypertriglyceridemia, omega-3 fatty acid supplements, such as Omacor, markedly reduce triglyceride levels by decreasing VLDL-C synthesis[3]
. An increase in LDL-C is sometimes seen with treatment[29]
. High doses of Omacor (2–4g daily) are needed to lower triglycerides, and lower doses are no longer recommended for the secondary prevention of cardiovascular events after myocardial infarction. Omacor is contraindicated where there is known hypersensitivity to the active substance, to soya or to any of the excipients[29]
.
PCSK9 inhibitors
Essential to the removal of LDL-C from the circulation is the LDL receptor proprotein convertase subtilisin/kexin type 9 (PCSK9), a secreted protease that has a role in controlling the number of LDL receptors available by binding LDL-C receptors and preventing them being expressed on the cell surface. Therefore, high levels of PCSK9 suppress LDL-C receptor activity by reducing the number of available receptors, which results in raised circulating cholesterol. Inhibition of PCSK9 will facilitate increased LDL-C receptor activity through an increased number of receptors and therefore lower circulating cholesterol levels[30],[31],[32]
.
Alirocumab and evolocumab, both monoclonal antibody PCSK9 inhibitors, have recently been licensed as options in the management of primary FH and non-familial hypercholesterolaemia in specific circumstances. In the studies to date, evolocumab with or without background statin therapy has been shown to reduce LDL-L levels by between 55% and 75% compared with placebo, and by up to 50% more than ezetimibe[33],[34],[35]
. Across five clinical trials involving 2,476 patients with familial hyperlipidaemia on optimal statin therapy, the average reduction in LDL cholesterol with alirocumab ranged from 36% to 59% compared with placebo[36]
. Cardiovascular outcome studies with both evolocumab and alirocumab are ongoing and are due to report from 2017. NICE has recently published the final appraisal determination for evolocumab (final NICE technology appraisal due in June 2016) and it states that evolocumab is recommended as an option for treating primary hypercholesterolaemia or mixed dyslipidaemia, only if:
- The dosage is 140mg every two weeks;
- LDL concentrations are persistently above the thresholds specified in ‘Table 4: Low density lipoprotein cholesterol concentrations above which evolocumab and alirocumab are recommended’ despite maximal tolerated lipid-lowering therapy. That is, either the maximum dose has been reached, or further titration is limited by intolerance (as defined in NICE’s guideline on FH: identification and management);
- The company provides evolocumab with the discount agreed in the patient access scheme[37]
.
Similarly, NICE has recently published the final appraisal determination for alirocumab (final NICE technology appraisal due in June 2016) and it states that alirocumab is recommended as an option for treating primary hypercholesterolaemia or mixed dyslipidaemia, only if:
- LDL concentrations are persistently above the thresholds specified in ‘Table 4: Low density lipoprotein cholesterol concentrations above which evolocumab and alirocumab are recommended’ despite maximal tolerated lipid-lowering therapy. That is, either the maximum dose has been reached or further titration is limited by intolerance (as defined in NICE’s guideline on FH: identification and management);
- The company provides alirocumab with the discount agreed in the patient access scheme[38]
.
| Table 4: Low density lipoprotein cholesterol concentrations above which evolocumab and alirocumab are recommended | ||||
|---|---|---|---|---|
| Without CVD | With CVD | |||
| High risk of CVD* | Very high risk of CVD** | |||
| Evolucumab and alirocumab | Primary non-familial hypercholesterolaemia or mixed dyslipidaemia | Not recommended at any LDL-C concentration | Recommended only if LDL-C concentration is persistently above 4.0mmol/L | Recommended only if LDL-C concentration is persistently above 3.5mmol/L |
| Primary heterozygous-familial hypercholesterolaemia | Recommended only if LDL-C concentration is persistently above 5.0mmol/L | Recommended only if LDL-C concentration is persistently above 3.5mmol/L | ||
Adapted from NICE guidance[37],[38] *High risk of CVD is defined as a history of any of the following: acute coronary syndrome (such as myocardial infarction of unstable angina needing hospitalisation); coronary or other arterial revasclarisation procedures; chronic heart disease; ishaemic stroke; peripheral arterial disease. **Very high risk of CVD is defined as recurrent cardiovascular events or cardiovascular events in more than one vascular bed (that is polyvascular disease). | ||||
Both drugs are administered by subcutaneous injection and require storage in a refrigerator. Both are contraindicated in patients with known hypersensitivity to the active ingredient or excipients. In the trials to date, both drugs have been well tolerated and the most common side effects were symptoms of influenza, nasopharyngitis, upper respiratory tract infection, rash, nausea, backache, arthralgia and injection site reactions[39],[40]
. For maintenance doses of PCSK9 inhibitors used in clinical practice, see ‘Table 5: PCSK9 inhibitors prescribed in clinical practice’.
| Table 5: PCSK9 inhibitors prescribed in clinical practice | |
|---|---|
| Usual maintenance dose | |
| Alirocumab[40] | 75–150mg every two weeks by subcutaneous injection |
| Evolucumab[39] | 140mg every two weeks or 420mg every month by subcutaneous injection |
PCSK9 inhibitors are marketed at a list price of approximately £4,000–4,500 per annum but NICE guidance indicates that a discounted price is available to the NHS through a patient access scheme[37],[38]
. For a summary of the side effects, drug interactions and monitoring of lipid-lowering drugs discussed in this article, see ‘Table 6: Side effects, drug interactions and monitoring of lipid-lowering drugs’.
| Table 6: Side effects, drug interactions and monitoring of lipid-lowering drugs | |||
|---|---|---|---|
| Drug | Side effects | Drug interactions | Monitoring |
| Ezetimibe[45] | Upper respiratory tract infection, diarrhoea, arthralgia, sinusitis, pain in extremities. | Increased levels of cyclosporine and ezetimibe in concomitant use, cholestyramine reduces ezetimibe levels, may affect INR for patients on vitamin K antagonists, such as warfarin. | Lipid levels within three months of initiation and at least annually thereafter. |
| Fibrates[19],[20] | Nausea, diarrhoea and abdominal pain are common, particularly on initiation. Myositis associated with muscle pain, unusual tiredness or weakness. | Enhanced effect of oral anticoagulants, risk of muscle toxicity with statins, increased risk of kidney injury if used in combination with cyclosporine or other immunosuppressant. | Lipid levels within three months of initiation and at least annually thereafter. Renal function at intervals throughout therapy. |
| Bile acid binders [22],[23],[24] | Indigestion, bloating, nausea, constipation, abdominal pain, flatulence. | Reduced absorption of anionic drugs such as warfarin, beta blockers, digoxin, levothyroxine. | Lipid levels, including trigycerides, within three months of initiation and at least annually thereafter. |
| Nicotinic acid derivatives | Flushing, itching, tingling, headache, nausea, heartburn, fatigue, rash, worsening of peptic ulcer, elevation in serum glucose and uric acid, hepatitis and elevation in hepatic transaminase levels. | Hypotension with BP-lowering drugs such as α-blockers possible; diabetics taking insulin or oral agents may require dose adjustment because of an increase in serum glucose levels. | Lipid profile every four to eight weeks during dose titration phase and then at least annually once stable dose achieved. Liver function tests are taken at baseline and every six to eight weeks during dose titration, then as needed for symptoms. Uric acid and glucose at baseline and again after stable dose reached (or symptoms produced), more frequently in diabetic patients. |
| Omega-3 fatty acid supplements[29] | Gastrointestinal disorders, including abdominal distension, abdominal pain, constipation, diarrhoea, dyspepsia, flatulence, eructation, gastro-oesophageal reflux disease, nausea or vomiting. | Omacor may potentiate the effect of warfarin on INR. | Lipid profile within three months of initiation, after a dose change and at least annually thereafter. |
| PCSK9 inhibitors[40], [41] | Nasopharyngitis, injection site reactions, upper respiratory tract infection and influenza. | Statins and other lipid-modifying therapy are known to increase production of PCSK9 — this leads to increased clearance and reduced systemic levels of PCSK9 inhibitor. | Lipid profile within three months of initiation, after a dose change and at least annually thereafter. |
| Adapted from Circulation [41] | |||
Alternative treatment options
Lipoprotein apheresis
Patients with severe treatment-resistant hypercholesterolaemia can be treated with lipoprotein apheresis – currently approximately 70 patients are receiving this treatment in England and Wales[42]
. Lipoprotein apheresis removes LDL-C, lipoprotein(a) and triglycerides from the plasma. This treatment can lower LDL-C by 75%, but the effect is transient and treatment needs to be repeated at regular intervals[43]
. The efficacy and superiority of lipoprotein apheresis in comparison with standard lipid lowering therapies is well established, however, there is still uncertainty regarding the ability of lipoprotein apheresis to allow for atherosclerotic plaque regression, since its effects are only transient. Furthermore, although lipoprotein apheresis is a highly effective treatment, its uptake and application is restricted because it has limited availability at specialist centres and treatments are needed on a weekly or two-weekly basis[45]
.
NICE recommends that lipoprotein apheresis is considered for the management of people with clinical homozygous FH and also, in exceptional circumstances (e.g. progressive, symptomatic coronary heart disease despite maximal oral lipid-lowering therapy and optimal medical/surgical intervention), for those with heterozygous FH[5]
.
Lomitapide
The drug lomitapide inhibits microsomal triglyceride transfer protein (MTP), which plays a major role in the assembly and secretion of the lipoproteins in the intestine and liver. As a result, lomitapide inhibits the synthesis of triglyceride-rich chylomicrons in the intestine and very low-density lipoprotein (VLDL; the precursor of LDL) in the liver. This results in an overall reduction in circulating cholesterol. Lomitapide has been studied extensively in patients with homozygous FH and, at a dose of 1mg/kg/day, lowered LDL-C by 50.9% from baseline[44]
. It is licensed only for use in patients with homozygous FH and may have a role in preventing progression to lipoprotein apheresis or, in those already receiving apheresis, to reduce the frequency of sessions to minimise the impact on quality of life. It is an option for a small cohort of patients and, at a cost of over £200,000 per annum, usage will remain low.
Lomitapide is initiated at a low 5mg dose that is increased gradually every four weeks to a maximum of 60mg daily.
It is contraindicated in:
- Patients with hypersensitivity to the active substance or any excipients;
- Patients with moderate or severe hepatic impairment and those with unexplained persistent abnormal liver function tests;
- Patients with a known significant or chronic bowel disease, such as inflammatory bowel disease or malabsorption, where there is concomitant administration of >40mg simvastatin or strong or moderate cytochrome P450 (CYP) 3A4 inhibitors (e.g. antifungal azoles, such as itraconazole, fluconazole, ketoconazole, voriconazole, posaconazole; macrolide antibiotics, such as erythromycin or clarithromycin; ketolide antibiotics, such as telithromycin; HIV protease inhibitors; the calcium channel blockers diltiazem and verapamil; the antiarrhythmic dronedarone);
- Pregnancy.
Adverse events that were noted during clinical studies included gastrointestinal symptoms (mainly diarrhoea), abnormal liver function tests (increased levels of aspartate transaminase and/or alanine aminotransferase and alkaline phosphatase), and increased hepatic fat content and possible steatohepatitis, the long-term consequences of which are unknown (e.g. progression to fibrosis, development of diabetes)[46]
.
Drugs in development
Cholesterylester transfer protein, which normally transfers cholesterol from HDL cholesterol to very low density or low density lipoproteins (VLDL or LDL), is inhibited by CETP inhibitors. Inhibition of CETP results in increased HDL levels and reduced LDL levels as cholesterol is moved from the peripheral tissues to the liver for excretion or recycling[47]
. In epidemiological studies, higher circulating levels of HDL have been associated with reduced cardiovascular events, hence the CETP inhibitors were thought to be a promising therapeutic option. However, of the original four CETP inhibitors in development, three have been abandoned because of increased mortality (torcetrapib) or lack of impact on cardiovascular events despite adequate HDL increases (dalcetrapib and evacetrapib)[48]
. Only anacetrapib remains in development with a large-scale cardiovascular outcomes study, REVEAL, due to report in 2017. The results of this study will determine any future role for CETP inhibitors, specifically anacretrapib, in clinical practice.
Summary
Once identified, statins remain the mainstay of therapy in most circumstances, but these alone are rarely sufficient to deliver the lipid lowering potency required in patients with primary dyslipidaemia. Other options, including ezetimibe, in line with NICE TA132, fibrates, bile acid binders, and omega-3 supplements, retain a role for the management of these conditions, usually under specialist supervision. The role of the PCSK9 inhibitors will be determined over the next 12–24 months, with cardiovascular outcomes data awaited. Lipoprotein apheresis and lomitapide remain options in a select number of patients with homozygous FH or, in the case of lipoprotein apheresis, for exceptional cases of heterozygous FH with an ongoing significant risk of cardiovascular events despite maximal treatment.
The preferred pharmacotherapy formulary for lipid management in FH and non-familial hypercholesterolaemia can be seen in ‘Figure 1: A preferred pharmacotherapy formulary for lipid management in familial and non-familial hypercholesterolaemia’.
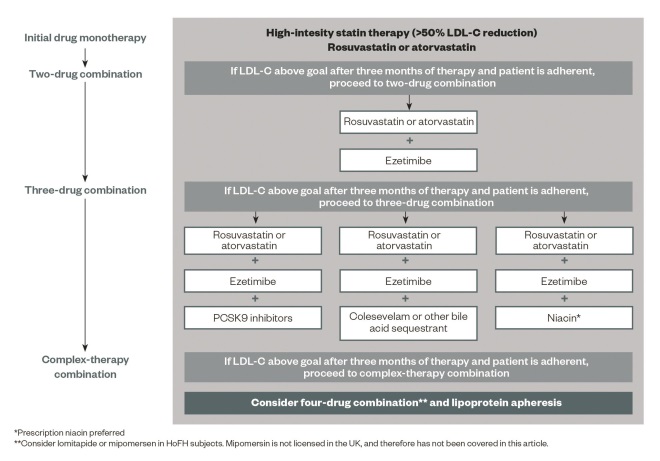
Figure 1: A preferred pharmacotherapy formulary for lipid management in familial and non-familial hypercholesterolaemia
Source: Reproduced with permission from Gidding SS, Champagne MA, de Ferranti SD et al. Circulation 2015;132
The decision to use a third-line agent must take into account many factors, including disease severity, patient reference, cost, and outcomes data. Future research on PCSK9 inhibitors and other new agents will inform the choice of a third agent, particularly in the context of statin intolerance.
Helen Williams is a consultant pharmacist for cardiovascular disease, south London, clinical associate for cardiovascular disease, Southwark CCG and clinical network lead for cardiovascular disease, Lambeth CCG. Correspondence to: helen.williams11@nhs.net
Author disclosure and conflicts of interest disclosure: The author has received speaker honoraria from and attended an advisory board for Merck Sharpe and Dohme, and received speaker honoraria from Amgen. No writing assistance was utilised in the production of this manuscript.
Reading this article counts towards your CPD
You can use the following forms to record your learning and action points from this article from Pharmaceutical Journal Publications.
Your CPD module results are stored against your account here at The Pharmaceutical Journal. You must be registered and logged into the site to do this. To review your module results, go to the ‘My Account’ tab and then ‘My CPD’.
Any training, learning or development activities that you undertake for CPD can also be recorded as evidence as part of your RPS Faculty practice-based portfolio when preparing for Faculty membership. To start your RPS Faculty journey today, access the portfolio and tools at www.rpharms.com/Faculty
If your learning was planned in advance, please click:
If your learning was spontaneous, please click:
References
[1] National Institute for Health and Care Excellence. Clinical Guideline 181 (2014). Lipid modification: cardiovascular risk assessment and the modification of blood lipids for the primary and secondary prevention of cardiovascular disease. Available at: http://www.nice.org.uk/guidance/cg181 (accessed May 2016).
[2] National Institute for Health and Care Excellence. Technology Appraisal Guideline 385 (2016). Ezetimibe for treating primary heterozygous-familial and non-familial hypercholesterolaemia. Available at: https://www.nice.org.uk/guidance/ta385 (accessed May 2016).
[3] Walker R & Williams H. Chapter 24: Dyslipidaemia in Walker R and Whittlesea. Clinical Pharmacy and Therapeutics. 5th Edition 2012. Churchill Livingstone: Elsevier.
[4] Fortson MR, Freedman SN & Webster PD 3rd. Clinical assessment of hyperlipidemic pancreatitis. Am J Gastroenterol 1995;90:2134. PMID: 8540502
[5] National Institute for Health and Care Excellence. Clinical Guideline 71 (2008). Identification and management of familial hypercholesterolaemia. Available at: http://www.nice.org.uk/guidance/cg71 (accessed May 2016).
[6] Benn M, Watts GF, Tybjaerg-Hansen A et al. Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab 2012;97(11):3956–3964. doi: 10.1210/jc.2012-1563
[7] Williams H. Dyslipidaemia: pathophysiology and types. Clinical Pharmacist 2013;5(1):189. doi: 10.1211/CP.2013.11125079
[8] Scientific Steering Committee on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ 1991;303(6807):893–896.
[9] Department of Health. Cardiovascular Disease Outcomes Strategy 2013. Available at: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/214895/9387-2900853-CVD-Outcomes_web1.pdf (accessed May 2016).
[10] Wierzbicki A. Professor in Cardiometabolic Disease. Personal communication. 6 May 2016.
[11] Kastelein JJP, Akdim F, Stroes ESG et al. for the ENHANCE investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008;358:1431–1443. doi: 10.1056/NEJMoa0800742
[12] Cannon CP, Blazing MA, Giugliano et al. for the IMPROVE-IT investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015;372:2387–2397. doi: 10.1056/NEJMoa1410489
[13] Robinson JG. Management of familial hypercholesterolaemia: a review of the recommendations from the National Lipid Association Expert Panel on familial hypercholesterolaemia. J Manag Care Pharm 2013;19:139–149. PMID: 23461430
[14] Frick MH, Elo O, Haapa K et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med 1987;317:1237–1245. doi: 10.1056/NEJM198711123172001
[15] Rubins HB, Robins SJ, Collins D et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med 1999;341:410–418. doi: 10.1056/NEJM199908053410604
[16] FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. The Lancet 2005;366(9500):1849–1861. doi: 10.1016/S0140-6736(05)67667-2
[17] The ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010;362:1563–1574. doi: 10.1056/NEJMoa1001282
[18] Pfizer. Lopid 300mg hard capsules. Summary of product characteristics. Available at: https://www.medicines.org.uk/emc/medicine/16878 (accessed May 2016).
[19] BGP Products. Lipantil Micro 200. Summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/684 (accessed May 2016).
[20] Actavis UK Ltd. Bezalip 200mg tablets. Summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/20856 (accessed May 2016).
[21] LRCCPP. The Lipid Research Clinics Coronary Primary Prevention Trial results: I. Reduction in incidence of coronary heart disease. JAMA 1984;251:351–364. doi: 10.1001/jama.1984.03340270029025
[22] Bristol Myers Squibb Holdings. Questran Powder for Oral Suspension 4g. Summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/23782 (accessed May 2016).
[23] Pfizer. Colestid granules for oral suspension summary of product characteristics. Available at: https://www.medicines.org.uk/emc/medicine/16796 (accessed May 2016).
[24] Sanofi. Cholestagel 625mg film coated tablets summary of product characteristics. Available at: https://www.medicines.org.uk/emc/medicine/20298 (accessed May 2016).
[25] Kamanna VS, Ganji SH & Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract 2009;63(9):1369–1377. doi: 10.1111/j.1742-1241.2009.02099.x
[26] European Medicines Agency. Acipimox only to be used as additional or alternative treatment to reduce high triglyceride levels. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/referrals/Nicotinic_acid/human_referral_prac_000020.jsp&mid=WC0b01ac05805c516f (accessed May 2016).
[27] HPS2-THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high risk patients. N Eng J Med 2014;371:203–212. doi: 10.1056/NEJMoa1300955
[28] European Medicines Agency. European Medicines Agency confirms recommendation to suspend Tredaptive, Pelzont and Trevaclyn. Press release: 18 January 2013. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2013/01/news_detail_001694.jsp&mid=WC0b01ac058004d5c1 (accessed May 2016).
[29] BGP Products. Omacor. Summary of product characteristics. Available at: https://www.medicines.org.uk/emc/medicine/10312 (accessed May 2016).
[30] Qian YW, Schmidt RJ, Zhang Y et al. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J Lipid Res 2007;48:1488–1498. doi: 10.1194/jlr.M700071-JLR200
[31] Horton JD, Cohen JC, Hobbs HH et al. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res 2009;50:S172–177. doi: 10.1194/jlr.R800091-JLR200
[32] Zhang DW, Lagace TA, Garuti R et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem 2007;282:18602–18612. doi: 10.1074/jbc.M702027200
[33] Robinson JG, Nedergaard BS, Rogers WJ et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA 2014;311:1870–1883. doi: 10.1001/jama.2014.4030
[34] Stroes E, Colquhoun D, Sullivan D et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol 2014;63:2541–2548. doi: 10.1016/j.jacc.2014.03.019
[35] Raal FJ, Stein EA, Dufour R et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. The Lancet 2015;385:331–340. doi: 10.1016/S0140-6736(14)61399-4
[36] FDA. FDA approves Praluent to treat certain patients with high cholesterol. Press release: 24 July 2015. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm455883.htm (accessed May 2016).
[37] National Institute for Health and Care Excellence. Final appraisal determination Evolocumab for treating primary hypercholesterolaemia and mixed dyslipidaemia. 2016. Available at: https://www.nice.org.uk/guidance/GID-TAG498/documents/final-appraisal-determination-document (accessed May 2016).
[38] National Institute for Health and Care Excellence. Final appraisal determination Alirocumab for treating primary hypercholesterolaemia and mixed dyslipidaemia. 2016. Available at: https://www.nice.org.uk/guidance/GID-TAG512/documents/final-appraisal-determination-document (accessed May 2016).
[39] Sanofi. Praluent Solution for infection. Summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/30956 (accessed May 2016).
[40] Amgen. Repatha Prefilled syringe. Summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/30627 (accessed May 2016).
[41] Gidding SS, Champagne MA, de Ferranti SD et al. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation 2015;132:2167-2192. doi: 10.1161/CIR.0000000000000297
[42] Heart-UK. LDL apheresis treatment FAQs 2015. Available at: http://heartuk.org.uk/statins-and-treatments/ldl-apheresis/ldl-apheresis-treatment-faqs (accessed May 2016).
[43] Thompson GR, Maher VMG, Kitano Y et al. Familial Hypercholesterolaemia Regression Study: a randomised trial of low-density-lipoprotein apheresis. The Lancet 1995;345:811–816. doi: 10.1016/S0140-6736(95)92961-4
[44] Cuchel M, Bloedon LT, Szapary PO et al. Inhibition of microsomal triglycerides transfer protein in familial hypercholesterolemia. N Engl J Med 2007;356:148–156. doi: 10.1056/NEJMoa061189
[45] Merck Sharpe & Dohme. Ezetrol 10mg tablets. Summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/12091 (accessed May 2016).
[46] Aegerion Pharmaceuticals Limited. Lojusta hard capsules. Summary of product characteristics. Available at: https://www.medicines.org.uk/emc/medicine/28321 (accessed May 2016).
[47] Hovingh GK, Davidson MH, Kastelein JJP et al. Diagnosis and treatment of familial hypercholesterolaemia. Eur H Journal 2013;34:962–971. doi: 10.1093/eurheartj/eht015
[48] Durrington PN. Cholesterol ester transfer protein (CETP) inhibitors. Br J Cardiol 2012;19:126–133. doi: 10.5837/bjc.2012.024