

Gordon Klein, Roger Bick, Brian Poindexter, UT Medical School / Science Photo Library
Around 550,000 people in the UK are recorded as having heart failure (HF), which equates to a prevalence of 0.8%[1]
. The true number (including those with unrecorded/undiagnosed HF) is likely to be much higher and estimated to be 920,000 in the UK[2]
. Depending on the definition applied, it is estimated that the prevalence of HF is around 1–2% of the adult population, increasing to over 10% in people aged over 70 years[3]
. Males are affected slightly more than females, with an estimated lifetime risk of HF at the age of 55 years being 33% for men compared with 28% for women[4]
.
Data from the 2015–2016 National Heart Failure Audit, which is carried out each year by the National Institute for Cardiovascular Outcomes Research, highlights that patient prognosis remains poor; mortality in patients admitted with HF is 8.9% during hospital admission, with a third of those discharged dying within the following year. Following admission to hospital for HF, survival rates are similar to those of colon cancer, and worse than those of breast or prostate cancer.
HF is the cause of, or complicates, around 5% of all emergency admissions in adults and consumes up to 2% of total NHS expenditure[5]
. Unfortunately, it is predicted that hospital admissions resulting from HF will continue to rise, mainly driven by an ageing population[2]
This article describes what happens to the heart during HF and how to treat it, as well as how to determine ejection fraction (EF).
Definition
HF is a complex clinical syndrome whereby the heart is unable to meet the metabolic demands of the body. Reasons for this mismatch can be multifactorial, and various underlying pathological conditions make the management and treatment complex. Initially, patients may tolerate small declines in output as various compensatory measures are activated to redress the balance. Over time, however, these compensatory measures become deleterious to heart function and lead to worsening cardiac output, resulting in further activation and additional stress on a failing heart.
Given the multitude of causes of HF, it is difficult to give a precise classification and definition compared with other common cardiac diseases, such as atrial fibrillation (AF), ischaemic heart disease (IHD) or hypertension, where a diagnosis is relatively straightforward. However, the European Society of Cardiology defines HF as a clinical syndrome characterised by symptoms such as shortness of breath, persistent coughing or wheezing, ankle swelling and fatigue, which may be accompanied by the following signs: elevated jugular venous pressure; pulmonary crackles; increased heart rate; and peripheral oedema[3]
. In addition, patients with HF often have several pathologies that may potentiate HF progression and symptoms. Diagnosis relies on clinical judgement based on a combination of patient history, physical examination and appropriate investigations to determine the likely underlying pathophysiology. Identification of the cause of HF is essential in the diagnostic workup, as this may offer specific therapeutic opportunities (i.e. if HF results from valvular causes, surgery may be curative).
Symptom classification
HF is a clinical syndrome characterised by a number of typical symptoms, such as breathlessness, oedema and fatigue. HF can be categorised in a number of different ways, although the underlying aetiologies are numerous. It is common practice in the UK to use the New York Heart Association (NYHA) functional classification to document and assess severity of symptoms and physical activity (see Table 1). It is also common to differentiate between left and right-sided HF, although the failure of one side often leads to failure of the other (biventricular failure). This is because the circulatory system works on a double pump with blood from the left ventricle being pumped systemically, returning to the right side where it is pumped towards the lungs for oxygenation; blood then returns to the left side for distribution round the systemic circulation again. If there is a failing on one side, given they are linked, failure on the other side will often follow.
| Table 1: New York Heart Association functional classification | |
| Class I | No limitation of physical activity. Ordinary physical activity does not cause undue breathlessness, fatigue or palpitations |
| Class II | Slight limitation of physical activity. Comfortable at rest, but ordinary physical activity results in undue breathlessness, fatigue or palpitations |
| Class III | Marked limitation of physical activity. Comfortable at rest, but less than ordinary physical activity results in undue breathlessness, fatigue or palpitations |
| Class IV | Unable to carry out any physical activity without discomfort. Symptoms at rest can be present. If any physical activity is undertaken, discomfort is increased |
| Source: Reproduced with permission from the American Heart Association. Classes of heart failure. 2017. Available at: http://www.heart.org/en/health-topics/heart-failure/what-is-heart-failure/classes-of-heart-failure (accessed December 2018) | |
A further differentiation can be made on whether there is dysfunction with the active pumping action in the ventricle (systolic dysfunction), or whether there is a problem with the relaxation or filling of the ventricle (diastolic dysfunction).
Diagnosis
Determining ejection fraction
The ‘gold standard’ tool for the assessment and diagnosis of HF is a transthoracic echocardiogram (TTE). This test allows a thorough assessment of chamber dimensions, valvular function and various estimates of heart function, including an estimation of EF (the proportion of blood ejected from the heart during a heartbeat). EF, which is typically between 55% and 70%, can be calculated using the following equation:
Ejection fraction = end diastolic volume – end systolic volume/end diastolic volume
HF is often classified based on the EF. HF with reduced ejection fraction (HFrEF) is identified where the EF is less than 40%. HF with preserved ejection fraction (HFpEF) can be harder to diagnose, and is defined as EF >50% with other markers of heart failure (e.g. diastolic dysfunction or structural heart disease). This important as underlying pathologies are distinct, with treatment in people with HFrEF having a strong evidence base in reducing mortality and morbidity. This is not replicated in patients with HFpEF, although patients typically exhibit similar clinical symptoms. Compared with HFrEF, patients with HFpEF are often older, more often female, and more commonly have a history of hypertension and valvular disease, while people with HFrEF tend to have a medical history that includes IHD[5]
. Table 2 represents the aetiology and comorbidity of patients with HF admitted to hospital in the UK.
Table 2: Aetiology and comorbidities of patients with heart failure | ||
| Medical history | HFrEF (%) | HFpEF (%) |
| Ischaemic heart disease | 48.4 | 37.9 |
| Atrial fibrillation | 49.1 | 40.0 |
| Myocardial infarction | 30.7 | 18.1 |
| Valve disease | 23.9 | 31.4 |
| Hypertension | 52.1 | 59.9 |
| Diabetes | 33.3 | 33.5 |
| Asthma | 8.4 | 9.4 |
| Chronic obstructive pulmonary disease | 16.7 | 18.9 |
| HFpEF: heart failure with preserved ejection fraction; HFrEF: heart failure with reduced ejection fraction Source: Reproduced with permission from the British Society for Heart Failure. National Heart Failure Audit, April 2015 to March 2016. Available at: http://www.ucl.ac.uk/nicor/audits/heartfailure/documents/annualreports/annual-report-2015-6-v8.pdf (accessed December 2018) | ||
To understand the differences and similarities in patients with HFrEF and HFpEF, it is important to understand another measure of heart function — cardiac output. Cardiac output can be defined as:
Cardiac output = stroke volume (amount of blood ejected in a heartbeat) x heart rate (beats per minute [bpm])
Generally, this is 70mL x 70bpm, which gives a cardiac output of 4.9L/min and the end diastolic volume is 110mL. Therefore, the EF will be 64% (110mL – 70mL / 110mL; see Figure 1A).
In a patient with HFrEF, stroke volume is typically reduced, which will therefore reduce cardiac output. For example, the patient’s stroke volume reduces to 44mL. This would result in an EF of 40% (44mL / 110mL) and a new cardiac output of 44mL x 70bpm = 3L/min (see Figure 1B).
In a patient with HFpEF, although the EF is normal, diastolic dysfunction is often present, lowering the end diastolic volume (either through ventricular hypertrophy, which reduces the cavity size, or through ventricular stiffening, where blood is not allowed to fill the ventricle). In either case, stroke volume ejected will be less because the initial volume is lower, which again leads to a reduction in stroke volume and a decline in cardiac output. For example, the patient’s end diastolic volume reduces to 70mL. As in the first case, the stroke volume is reduced to 44mL but the EF remains ‘normal’. This would result in an EF of 63% (44mL / 70mL) and a cardiac output of 3L/min (44mL x 70bpm; see Figure 1C).
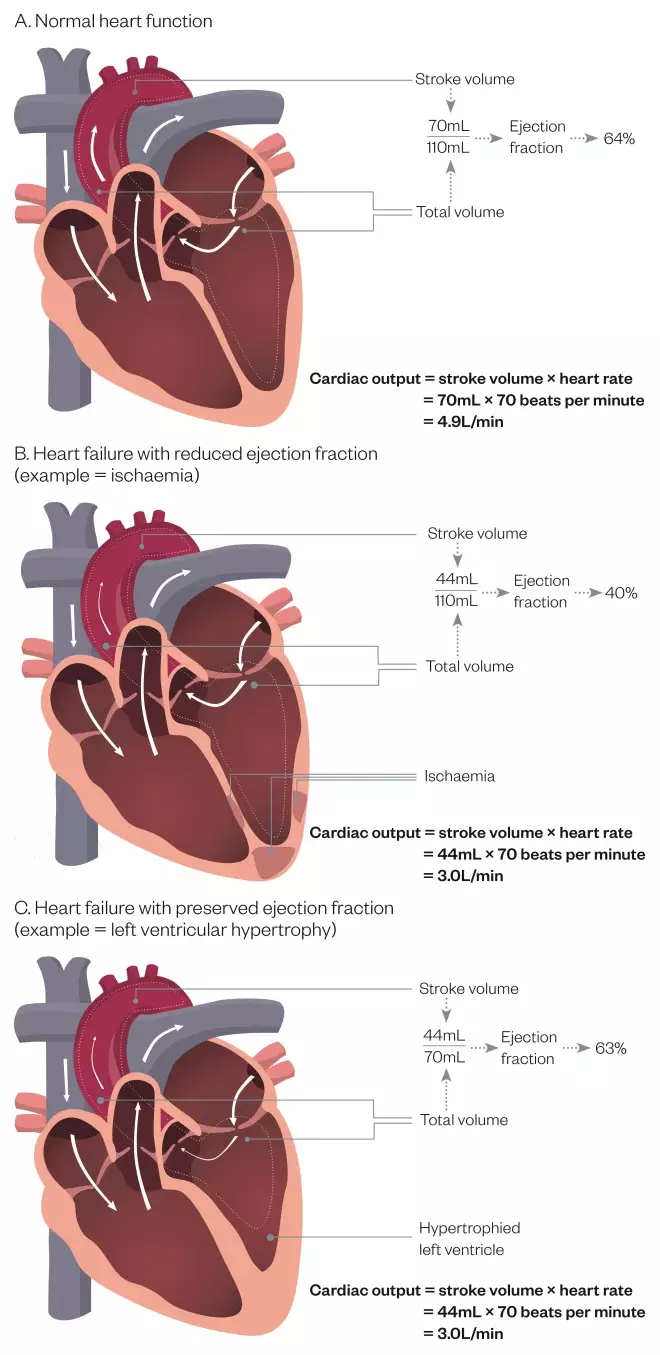
Figure 1: Representation of cardiac output in relation to ejection fraction
Source: Paul Wright & Martin Thomas
Biomarkers
Natriuretic peptides are released from the myocardium in response to ventricular wall stress, hypertrophy and fluid overload, representing useful biomarkers in the workup to HF diagnosis. B-type natriuretic peptide (BNP) and amino-terminal pro-B-type natriuretic peptide (NT-proBNP) are the two main biomarkers used clinically.
The heart continuously produces small amounts of a precursor proBNP, which is cleaved to release the active hormone BNP and an inactive fragment NT-proBNP. BNP and NT-proBNP are increased in patients with HF; therefore, low levels are useful to rule out HF. It should be noted that neprilysin inhibitors lead to an increase in circulating BNP and, therefore, BNP should not be used to monitor treatment or to assess response to treatment if neprilysin inhibitors are used. Instead, changes in NT-proBNP is still a useful reflection of the reduction in wall stress[6]
.
The latest National Institute for Health and Care Excellence (NICE) guidance for HF recommends measuring NT-proBNP in people with suspected HF[2]
. Since very high levels of NT-proBNP carry a poor prognosis, people with suspected HF and an NT-proBNP level >2,000 nanograms/L (236picomol/L) should be referred urgently for specialist assessment and TTE within two weeks. Patients with a level of 400–2,000 nanograms/L (47–236picomol/L) should also be referred for specialist assessment and TEE within six weeks. A NT-proBNP of <400 nanograms/L (47picomol/L) in an untreated person makes a diagnosis of HF less likely[2]
.
Various factors can affect circulating BNP and NT-proBNP levels and, therefore, caution must be taken when interpreting results (see Box 1).
Box 1: Factors that can affect circulating
B-type natriuretic peptide
and
amino-terminal pro-B-type natriuretic peptide
Factors that may lead to lower circulating levels:
- Obesity;
- African or Afro–Caribbean family origin;
- Treatment with:
- Diuretics;
- Angiotensin-converting enzyme inhibitors;
- Beta-blockers;
- Angiotensin II receptor blockers;
- Mineralocorticoid receptor antagonists.
Factors that can lead to higher circulating levels (other than heart failure):
- Age over 70 years;
- Left ventricular hypertrophy;
- Ischaemia;
- Tachycardia;
- Right ventricular overload;
- Hypoxaemia (including pulmonary embolism);
- Kidney dysfunction (estimated glomerular filtration rate <60mL/minute/1.73m2);
- Sepsis;
- Chronic obstructive pulmonary disease.
Source: National Institute for Health and Care Excellence. Chronic heart failure in adults: diagnosis and management [NG106]. 2018. Available at: https://www.nice.org.uk/guidance/ng106/resources/chronic-heart-failure-in-adults-diagnosis-and-management-pdf-66141541311685 (accessed December 2018)
Impact of compensatory mechanisms
A reduction in cardiac output in both HFrEF and HFpEF leads to compensatory mechanisms being activated to redress the imbalance. Initially, these may improve cardiac output but they become detrimental in the longer term as they place excessive strain on the failing heart. The main systems activated in response to a reduced systemic perfusion are the renin-angiotensin-aldosterone system (RAAS), sympathomimetic nervous system (SNS) and natriuretic peptide system (NPS). Other systems, such as parasympathetic activation, vasopressin, endothelin and other pathways, are outside the scope of this article (most therapeutic strategies to date are aimed at manipulating RAAS, SNS or NPS).
A reduction in cardiac output leads to reduced renal perfusion, which stimulates release of renin in the kidneys. This stimulates conversion of angiotensinogen from the liver to angiotensin I. This is promptly converted to angiotensin II on the surface of pulmonary and renal endothelium. The actions of angiotensin II include activation of the sympathomimetic pathway, arteriolar vasoconstriction and cardiac remodeling. Release of aldosterone leads to increased salt and water retention. Activation of the RAAS leads to increased circulating blood volume, which will increase pre-load with the aim to increase atrial pressure and, ultimately, increase the stroke volume to improve cardiac output[7]
.
The SNS is activated through release of catecholamines, such as noradrenaline and adrenaline. Activation of beta 1, beta 2 and alpha receptors aim to raise the heart rate and force of contraction with the aim to increase cardiac output. In addition, chronic activation leads to stimulation of the RAAS system to further increase pre-load[6]
.
Natriuretic peptides (NPs) have a wide range of effects on the kidneys, heart and central nervous system. NPs are released in response to atrial and ventricular stretch and act to oppose the effects of angiotensin II on systemic vascular resistance, vascular tone, aldosterone and cardiac remodeling. Activation of this system aims to counteract the effects of SNS and RAAS (see Figure 2). In addition, levels of NPs can be used to facilitate a diagnosis of HF as they are released when the heart is under strain[8]
.
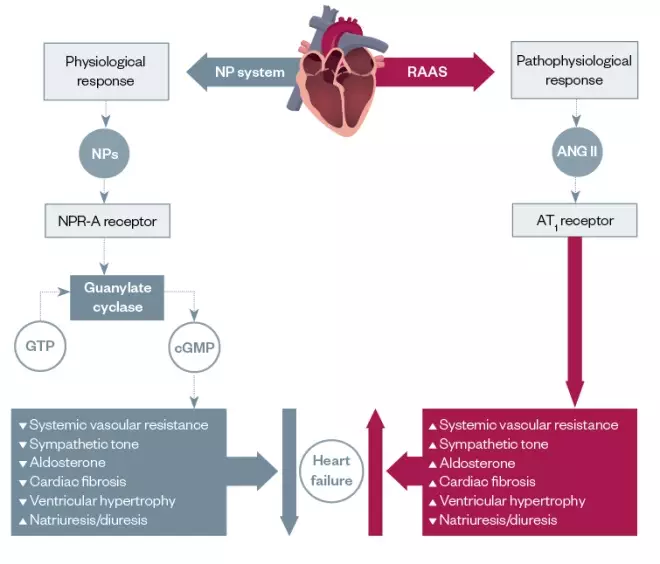
Figure 2: Activation of the renin angiotensin aldosterone system system
Source: Source: Drug Discovery Today: Therapeutic Strategies
[8]
ANG II: angiotensin II; AT1: angiotensin type 1; cGMP: cyclic guanosine monophosphate; GTP: guanosine-5’-triphosphate; NP: natriuretic peptide; NPR-A: neprilysin receptor A; RAAS: renin-angiotensin-aldosterone system
The Frank–Starling law of the heart states that stroke volume increases in response to an increase in volume of blood filling the heart (end diastolic volume) when all other factors remain constant. This adaptive mechanism is crucial for normal physiological function and allows rapid response to increased demands (e.g. when exercising). In chronic HFrEF, the myocardium with impaired contraction is no longer able to increase the force of contraction with increasing end diastolic volume and activation of RAAS and SNS become counterproductive, as they lead to increased cardiac stress and pre-load with the heart being unable to increase cardiac output.
Some have compared the Frank–Starling law with an analogy to an elastic band — as you pull the elastic band further apart, the recoil is greater, but if over-stretched, the elastic band can become weakened and you no longer get the recoil desired.
In HFpEF, the rise in left ventricular end diastolic pressure needed during exercise through accommodation of more pre-load cannot be sustained before the left ventricular end diastolic pressure rises to levels that usually result in significant rise in left atrial pressures, and pulmonary venous pressure resulting in the development of pulmonary congestion and breathlessness. This inability to increase cardiac output as the myocardium is unable to stretch in the first instance, leads to the expression ‘stiff ventricle’, which is frequently used to describe the physiology in HFpEF.
Depending on the underlying causes of HF, other adaptive processes can occur. Left ventricular hypertrophy (LVH), which is an enlargement and thickening of the walls of the heart, can occur in response to consistently raised ventricular pressure. This can commonly occur owing to persistent high blood pressure or in response to aortic stenosis. As LVH progresses, the increasing muscle mass reduces the end diastolic volume and will inevitably reduce the cardiac output, while the EF remains “normal”.
Decompensated heart failure
Also known as congestive HF, decompensated HF occurs in patients where there is increased blood pressure resulting from weak pump in HFrEF and stiff pump in HFpEF. In either case, there is a backfill of pressure in the lungs as the ventricle cannot eject enough blood out of the left ventricle. This leads to increased pulmonary pressure that pushes fluid out of capillaries and into aveolar space (causing pulmonary oedema). This additional fluid in the aveolar spaces slows diffusion of oxygen and carbon dioxide, leading to dyspnoea (shortness of breath).
Orthopnoea can also be present in decompensated HF; this is a feeling of breathlessness when lying flat, which is caused as more venous blood flows back into the heart and into pulmonary circulation when the patient assumes a supine posture, again leading to raised pulmonary pressures and migration of fluid into aveolar space. Rales, or ‘crackles’, are heard when listening to the chest through a stethoscope, at least in the lung bases. Similar to the mechanism of pulmonary congestion, when there is either a rise in the pulmonary artery pressure and/or impairment of the right ventricle, back pressure would lead to fluid accumulation in the liver, legs, sacrum and peritoneal cavity, causing ascites.
Treatment
Diuretics to reduce congestion
Diuretics are used for HF of all causes to reduce congestion. While they relieve symptoms associated with HF, they are generally regarded as not affecting the underlying disease pathophysiology. In clinical practice, careful management of a patient’s fluid status with diuretics can prevent or delay admission to hospital with acute decompensated HF. This finding is echoed in a meta-analysis of diuretic use in congestive HF, suggesting a mortality benefit with their use and a reduction of admission[9]
.
In the UK, furosemide is commonly used as a first-line loop diuretic. Doses will be adjusted according to symptoms and clinical status. Clinical examination of peripheral oedema, pulmonary oedema and jugular venous pressure are used in this assessment. Typically, doses of 40–80mg daily are used with doses up to 80mg twice daily in more severe cases. Bumetanide, another loop diuretic, has a higher and more consistent bioavailability than furosemide (>90%), whose bioavailability ranges from 10–90% with food intake delaying absorption and reducing peak concentration[10]
.
In patients who are congested, absorption through an oedematous gastrointestinal tract is affected less with bumetanide than with furosemide use, and some patients respond more consistently to oral bumetanide than to furosemide.
In cases of diuretic resistance (i.e. lack of clinical effect despite large doses of loop diuretic), a thiazide (e.g. bendroflumethiazide) or thiazide-like diuretic (e.g. metolazone, although this is unlicensed in the UK) can be added that act synergistically to offload fluid. In general, the response to adding metolazone is more potent than bendroflumethiazide, especially when there is reduced kidney function. Therefore, much higher doses of bendroflumethiazide are needed, whereas metolazone continues to work well even at lower estimated glomerular filtration rates. Typically, doses of 2.5mg are used as a single dose or on alternate days. Doses can be increased up to response or to 10mg daily, although electrolyte disturbance generally means daily doses should not exceed 5mg. Owing to the potency of this combination and the likelihood of kidney dysfunction and electrolyte disturbance, this combination should be monitored very frequently with long-term use rarely seen in clinical practice.
Aggressive use of diuretics can cause a dramatic reduction of circulating blood volume that can lead to hypotension and kindey impairment. Patients should aim for weight loss not to exceed more than 0.5–1.0kg a day while being treated with high-dose diuretics. Electrolyte loss is also common with high-dose diuretics, which can limit their use, particularly hyponatraemia (which can be difficult to treat). It should be noted that patients who have a significant overload of fluid may have dilutional hyponatraemia, which is a poor marker of response and can be difficult to treat. Failure to respond to combinations of loop and thiazide diuretics, or hyponatraemia with accompanying fluid gain, would warrant advanced HF input where other strategies such as intravenous diuresis, high-dose mineralocorticoid receptor antagonists or extracorporeal ultrafiltration may be considered.
Treatment for heart fraction with preserved ejection fraction
No treatment has yet been shown to reduce morbidity or mortality in patients with HFpEF. Management of HFpEF is directed towards managing the underlying comorbidities (e.g. hypertension, IHD and diabetes) alongside diuretic use to manage symptoms of fluid overload[2]
.
Pharmacological management of heart failure with reduced ejection fraction
There is clear evidence that initiation of disease-modifying drugs in HFrEF can lead to significant reductions in mortality and morbidity. According to the National Heart Failure Audit Report 2015–2016, patients admitted to hospital with left ventricular systolic dysfunction that survived and were discharged with no prognostic medication had a one-year mortality rate of 49.3%, compared with those that were discharged on an angiotensin-converting enzyme inhibitor (ACEI) or angiotensin receptor blockers (ARBs), and beta blockers (BBs) and mineralocorticoid receptor antagonists (MRAs) who had a one-year mortality rate of 23.4%[5]
. This demonstrates the real-world value in ensuring patients are medically optimised. Estimates for the individual reduction in mortality associated with the disease-modifying agents are in HFrEF are presented in Figure 3.
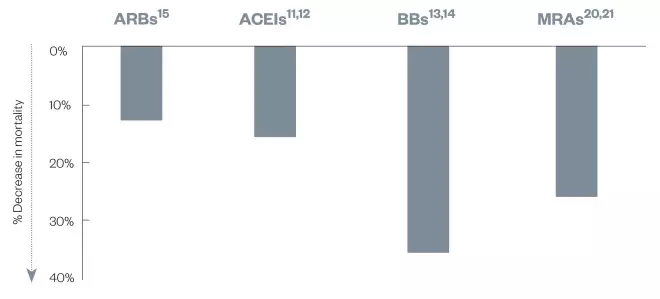
Figure 3: Drugs that reduce mortality in heart failure with reduced ejection fraction
Source: SOLVD
[11], AIRE
[12], MERIT
[13], CIBIS-II
[14], CHARM
[15], RALES
[20], EMPHASIS
[21]
ACEI: angiotensin-converting enzyme inhibitor; ARB: angiotensin receptor blocker; BB: beta blocker; MRA: mineralocorticoid receptor antagonist
The recently updated NICE guidance for HF includes a treatment algorithm (see Figure 4); the following sections summarise this guidance[2]
.
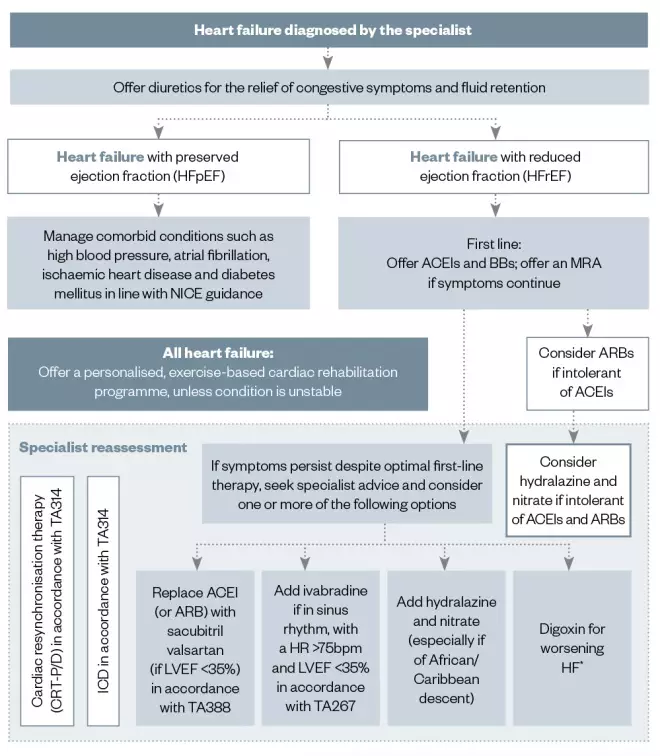
Figure 4: National Institute for Health and Care Excellence therapeutic algorithm
*Please refer to CG180 for recommendations on the use of digoxin in patients with atrial fibrillationACEI: angiotensin-converting enzyme inhibitor; ARB: angiotensin receptor blocker; BB: beta blocker; bpm: beats per minute; CG180: NICE Clinical Guidance 180; CRT-P/D: cardiac resynchronisation therapy — pacing/defibrillator; eGFR: estimated glomerular filtration rate; HR: heart rate; LVEF: left ventricular ejection fraction; MRA: mineralocorticoid receptor antagonist; NICE: National Institute of Health and Care Excellence; TA267: NICE Technology Appraisal 267; TA314: NICE Technology Appraisal 314; TA388: NICE Technology Appraisal 388
If the person’s eGFR is 30–45mL/min/1.73m2, consider lower doses and/or slower titration of dose of ACEIs or ARBs, MRAs, sacubitril valsartan and digoxin
If the person’s eGFR is <30mL/min/1.73m2, consider liaising with a renal physician
ACEIs together with BBs are considered first-line agents in the treatment of HFrEF, and both have been demonstrated to reduce morbidity and mortality[11],[12],[13],[14]
. Both agents should be titrated up to maximum tolerated doses in order to replicate results from the large randomised controlled trial that demonstrated their benefits. Where ACEIs are not tolerated, an ARB can be considered[15]
. Despite the strong evidence base, in clinical practice the majority of patients receive sub-optimal doses of ACEIs and BBs, with around 30% of patients reaching target doses[16],[17]
.
Specific instructions for titration may vary slightly between different agents but a pragmatic approach to initiating ACEIs or ARBs in a community setting would be to start low and double the dose at no quicker than two-weekly intervals[2],[3]
. Up-titration of ACEIs should be continued, providing patients are tolerating this well and not suffering from symptomatic hypotension. It is essential that kidney function is checked prior to initiation and rechecked one to two weeks after and after any dose titrations to monitor for kidney dysfunction.
It has been suggested that in UK practice, only a tenth of patients initiating ACEI or ARB therapy receive the guideline recommended creatinine monitoring[18]
. In the community setting, there are useful resources, such as the Think Kidneys position statement that offers practical advice on monitoring and acting on changes to serum creatinine and potassium, and what to do in patients with HF[19]
.
BBs licensed for the treatment of HF can be initiated together with ACEIs at low doses and in a similar fashion, with doses doubled at intervals of no quicker than two weeks[2],[3]
. It is generally accepted that BBs should not be started in patients presenting with acute decompensated HF, but can be initiated in clinically stable patients. Given the side effects of BBs that can mimic symptoms of HF (particularly fatigue and, in some instances, shortness of breath), titration should adopt a low and slow approach. The maximum tolerated dose should be aimed for with both BBs and ACEIs. BBs should not be withheld because of age or the presence of peripheral vascular disease, erectile dysfunction, diabetes, interstitial lung disease or chronic obstructive pulmonary disease[2]
.
In practice, although patients with HFrEF are commonly prescribed prognostic ACEIs and BBs (a European registry suggested 92% and 93%, respectively), the proportion of patients reaching target doses was only 29% and 18%, respectively; this is in stark contrast to clinical trials in which 50–60% of patients achieve target doses[17]
.
The two available MRAs (spironolactone and eplerenone) are recommended in symptomatic patients with an EF < 35%; both agents demonstrate a relative reduction in mortality of around 30%[20],[21]
. As they are added to ACEIs or ARBs, there is concern of hyperkalaemia. Consequently, it is recommended to measure urea and electrolytes within one week of initiation, or up-titration of spironolactone or epleronone monthly for the first three months, three-monthly for one year and four-monthly thereafter[18]
.
Angiotensin receptor neprilysin inhibitors
More recently, a better understanding of the neurohormonal activation and pathways has led to the successful development of a new class of agent in the management of patients with HFrEF. Sacubitril, a neprilysin inhibitor has been combined with valsartan (an ARB) to create a compound marketed as Entresto® (Novartis). Collectively, this compound is known as an angiotensin receptor neprilysin inhibitor.
On their own, neprilysin inhibitors have failed to demonstrate adequate clinical outcomes in HF, which is most likely a result of neprilysin inhibition leading to a rise in natriuretic peptide levels but also in angiotensin II levels. For this reason, inhibition of neprilysin and dual blockade of the RAAS system inhibitor was a logical next step in its development[6]
. A previous compound combining a neprilysin inhibitor with an ACEI (omapatrilat) was shown to be non-inferior to enalapril but was associated with an increased risk (albeit low absolute numbers) of life-threatening facial angioedema[22]
. For this reason, it is essential that sacubitril valsartan is not to be given within 36 hours after ACEI administration, likewise ACEIs are not be given within 36 hours after sacubitril administration. This is not the same for ARBs, where sacubitril valsartan can be initiated at the next due dose of ARB.
Sacubitril valsartan
In a large clinical trial where sacubitril valsartan was compared with enalapril, which is considered a gold standard treatment, the trial was stopped early owing to favourable effects of sacubitril valsartan. There was a 20% relative-risk reduction in the composite outcome of cardiovascular death or hospitilisation for HF and a 16% reduction in death from any cause. In addition, there were fewer heart failure symptoms in the sacubitril valsartan group with improvements in NYHA class[23]
. Despite this improvement, mortality rate from cardiovascular causes in the treatment arm was 13.3% over the course of the trial that had a median follow-up duration of 27 months.
NICE recommends that sacubitril valsartan is an option for treating symptomatic chronic HFrEF, only in patients:
- With NYHA class II to IV symptoms;
- With a left ventricular EF of 35% or less; and
- Who are already taking a stable dose of ACEI or ARB[24]
.
Treatment with sacubitril valsartan should be started by HF specialists with access to a multidisciplinary HF team. As recommended in the summary of product characteristics for sacuitril valsartan, it should not be initiated in patients with systolic blood pressure <100mmHg. In terms of monitoring, sacubitril valsartan can be thought of as being very similar to ACEIs or ARBs, with initiation requiring monitoring of kidney function and potassium in a similar manner as described above for ACEIs. However, it should be noted that this is poorly done with ACEIs and ARBs in general practice[17]
.
Additional therapies for heart failure
Additional medical therapies that may be considered for patients with HFrEF groups include ivabradine, digoxin and hydralazine in combination with isosorbide dinitrate. Use of these agents is usually under direct supervision of HF specialists when other therapies are not tolerated or in patients who remain symptomatic despite maximum tolerated titration of other agents. It is outside the scope of this article, but device therapies, such as cardiac resynchronisation therapy, can make significant symptomatic improvement in a selected cohort with HFrEF and implantable cardioverter defibrillators can reduce sudden cardiac death in appropriately selected patients.
Other considerations
AF is common to those presenting with HF. In part, this is owing to the prevalence of AF increasing sharply aged over 70 years. Patients in AF are more likely to present with HF as the coordinated atrial push of blood into the ventricles is lost, meaning cardiac output is likely to decline. The reduction in cardiac output can be as high as 30%, which can have a significant impact on patients whose hearts are already struggling owing to HF. A recent study has suggested that catheter ablation for AF in patients with HFrEF can lead to reductions in mortality and reduced admissions with HF[25]
. However, it remains to be seen if this trial will translate to wider clinical practice.
Optimisation of glucose control in patients with diabetes has been shown to reduce microvascular but not macrovascular complications. More recently, empagliflozin, an inhibitor of sodium–glucose cotransporter 2 (SGLT2), was the first to demonstrate a reduction of cardiovascular mortality. It was also noted to reduce hospitilisations for HF[26]
. While there are no results specifically looking at randomising patients with HF to a SGLT2 inhibitor, the signal that these agents may reduce hospitalisations and cardiovascular mortality is attractive, especially if patients are failing to achieve blood glucose goals.
Summary
There is likely to be a considerable increase in patients presenting with HF over the next decade, as the population ages. Pharmacists can help minimise the burden of HF, both in terms of NHS resources and patient morbidity and mortality, by not only ensuring that patients with HFrEF are optimised on maximum tolerated doses of evidence-based medication, but by helping patients implement appropriate lifestyle changes. These include smoking cessation, making dietary changes, losing weight, increasing physical activity (aiming for at least 30 minutes of exercise on most days of the week) and moderating alcohol intake to below recommended limits.
Patients should also be counselled around self-care principles, so that they are able to recognise worsening symptoms of dyspnoea, oedema, sudden weight gain and nocturnal symptoms as worsening HF symptoms.
There is still a long way to go in improving outcomes for patients with HF, despite the success of sacubitril valsartan. In addition, there is huge opportunity to improve dose titrations of prognostic HF medication as many patients, despite being on ACEIs, BBs and MRAs, are seldom titrated to evidence-based doses in practice.
Financial and conflicts of interest disclosure:
The authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. No writing assistance was used in the production of this manuscript.
Sponsored by:
Novartis provided financial support in the production of this content. Clinical Pharmacist retains sole editorial responsibility. CVM18-E025
References
[1] British Heart Foundation. Cardiovascular disease statistics 2018. Available at: https://www.bhf.org.uk/what-we-do/our-research/heart-statistics/heart-statistics-publications/cardiovascular-disease-statistics-2018 (accessed December 2018)
[2] National Institute for Health and Care Excellence. Chronic heart failure in adults: diagnosis and management. NICE guideline [NG106]. 2018. Available at https://www.nice.org.uk/guidance/ng106/resources/chronic-heart-failure-in-adults-diagnosis-and-management-pdf-66141541311685 (accessed December 2018)
[3] Ponikowski P, Voors AA, Anker SD et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2016;37(27):2129–2200. doi: 10.1093/eurheartj/ehw128
[4] Bleumink GS, Knetsch AM, Sturkenboom MCJM et al. Quantifying the heart failure epidemic: prevalence, incidence rate, lifetime risk and prognosis of heart failure The Rotterdam Study. Eur Heart J 2004;25(18):1614–1619. doi: 10.1016/j.ehj.2004.06.038
[5] British Society for Heart Failure. National Heart Failure Audit: April 2015 to March 2016. 2017. Available at: http://www.ucl.ac.uk/nicor/audits/heartfailure/documents/annualreports/annual-report-2015-6-v8.pdf (accessed December 2018)
[6] Jhund PS & McMurray JJV. The neprilysin pathway in heart failure: a review and guide on the use of sacubitril/valsartan. Heart 2016;102(17):1342–1347. doi: 10.1136/heartjnl-2014-306775
[7] Jackson G, Gibbs CR, Davies MK et al. ABC of heart failure. BMJ 2000;320:167–170. doi: 10.1136/bmj.320.7228.167
[8] Langenickel TH & Dole WP. Angiotensin receptor-neprilysin inhibition with LCZ696: a novel approach for the treatment of heart failure. Drug Discov Today Ther Strateg 2012;9(4):e131–139. doi: 10.1016/j.ddstr.2013.11.002
[9] Faris R, Flather M, Purcell H et al. Current evidence supporting the role of diuretics in heart failure: a meta analysis of randomised controlled trials. Int J Cardiol 2002;82(2):149–158. doi: 10.1016/S0167-5273(01)00600-3
[10] Ellison DH & Felker GM. Diuretic treatment in heart failure. NEJM 2017;377:1964–1975. doi: 10.1056/NEJMra1703100
[11] The SOLVD investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. NEJM 1991;325:293–302. doi: 10.1056/NEJM199108013250501
[12] The AIRE study investigators. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet 1993;342(8875):821–828. doi: 10.1016/0140-6736(93)92693-N
[13] MERIT HF study group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF) Lancet 1999;353(9169):2001–2007. doi: 10.1016/S0140-6736(99)04440-2
[14] CIBIS-II investigators and committees. The cardiac insufficiency bisoprolol study II (CIBIS II) a randomised trial. Lancet 1999;353(9146):9–13. doi: 10.1016/S0140-6736(98)11181-9
[15] Young JB, Dunlap ME, Pfeffer MA et al. Mortality and morbidity reduction with candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: results of the CHARM low-left ventricular ejection fraction trials. Circulation 2004;110(17):2618–2626. doi: 10.1161/01.CIR.0000146819.43235.A9
[16] Russell SJ, Oliver M, Edmunds L et al. Optimised beta blocker therapy in heart failure: is there space for additional heart rate control? Br J Cardiol 2012;19:21–23. doi: 10.5837/bjc.2012.001
[17] Maggioni AP, Anker SD, Dahlström U et al. Are hospitalised or ambulatory patients with heart failure treated in accordance with European Society of Cardiology guidelines? Evidence from 12,440 patients of the ESC Heart failure long-term registry. Eur J Heart Fail 2013;15(10):1173–1184. doi: 10.1093/eurjhf/hft134
[18] Schmidt M, Mansfield KE, Bhaskaran K et al. Adherence to guidelines for creatinine and potassium monitoring and discontinuation following renin-angiotensin system blockade; a UK general practice based cohort study. BMJ Open 2017;7(1):e012818. doi: 10.1136/bmjopen-2016-012818
[19] Think Kidneys, The Renal Association & The British Society for Heart Failure. Changes in kidney function and serum potassium during ACEi/ARB/diuretic treatment in primary care. 2017. Available at: https://www.thinkkidneys.nhs.uk/aki/wp-content/uploads/sites/2/2017/10/Changes-in-Kidney-Function-FINAL.pdf (accessed December 2018)
[20] Pitt B, Zannad F, Remme WJ et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. NEJM 1999;341:709–717. doi: 10.1056/NEJM199909023411001
[21] Zannad F, McMurray JJ, Krum H et al. Eplerenone in patients with systolic heart failure and mild symptoms. NEJM 2011;364:11–21. doi: 10.1056/NEJMoa1009492
[22] Packer M, Califf RM, Konstam ME et al. Comparison on omapatrilat and enalapril in patients with chronic heart failure: the omapatrilat versus enalapril randomized trial of utility in reducing events (OVERTURE). Circulation 2002;106:920–926. PMID: 12186794
[23] McMurray JJV, Packer M, Desai A et al. Angiotensin – neprilysin inhibition versus enalapril in heart failure. NEJM 2014;371:993–1004. doi: 10.1056/NEJMoa1409077
[24] National Institute for Health and Care Excellence. Sacubitril valsartan for treating symptomatic chronic heart failure with reduced ejection fraction. Technology appraisal guidance [TA388]. 2016. Available at: https://www.nice.org.uk/guidance/ta388/resources/sacubitril-valsartan-for-treating-symptomatic-chronic-heart-failure-with-reduced-ejection-fraction-pdf-82602856425157 (accessed December 2018)
[25] Marrouche NF, Brachmann J, Andresen D et al. Catheter ablation for atrial fibrillation with heart failure. NEJM 2018;378:417–427. doi: 10.1056/NEJMoa1707855
[26] Zinman B, Wanner C, Lachin JM et al. Empagliflozin, cardiovascular outcomes and mortality in type 2 diabetes. NEJM 2015;373:2117–2128. doi: 10.1056/NEJMoa1504720
You might also be interested in…

Once-weekly HIV pill performs as well as daily treatment in phase III study

Government ‘missing huge trick’ excluding pharmacies from NHS capital investment plan, say leaders
