PR. J. Bernard / CNRI / Science Photo Library
Cancer learning ‘hub’
Pharmacists are playing an increasingly important role in supporting patients with cancer, working within multidisciplinary teams and improving outcomes. However, in a rapidly evolving field with numbers of new cancer medicines is increasing and the potential for adverse effects, it is now more important than ever for pharmacists to have a solid understanding of the principles of cancer biology, its diagnosis and approaches to treatment and prevention. This new collection of cancer content, brought to you in partnership with BeOne Medicines, provides access to educational resources that support professional development for improved patient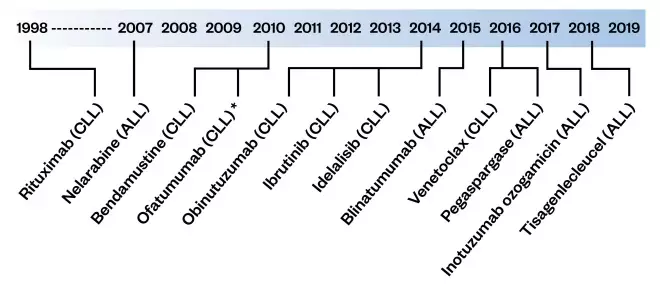
Figure 1: Timeline of European Medicines Authority approval dates for treatment for acute lymphoblastic leukaemia (ALL) and chronic lymphocytic leukaemia (CLL)
ALL: acute lymphoblastic leukaemia; CLL: chronic lymphoblastic leukaemia *Withdrawn on 28 February 2019
Acute lymphoblastic leukaemia
Incidence
LL is the most common cancer in children, with around 400 new cases of childhood leukaemia in the UK each year — 75% of these are ALL[1]. It can affect children of any age, but is most often seen in children aged between one and four years old. In adults, ALL is much less common, with an annual incidence of around 1 per 100,000 people; however, a similar total number of new diagnoses are seen in the paediatric setting each year[2].Aetiology
The exact cause of ALL is unknown, although there are certain risk factors that mean some patients are more at risk than others. For example, children with certain genetic disorders, such as Down’s syndrome (DS), are known to have a higher risk of developing leukaemias[1]. In 2018, it was proposed that development of ALL in children is a two-stage process: an in utero, genetic mutation leads to the development of a pre-leukaemic clone; and a second, postnatal genetic change — putatively triggered by infection — leads to the development of the observable disease[3].Presentation and diagnosis
As leukaemia cells multiply in the bone marrow, the production of normal blood cells is reduced. The presenting symptoms arise from this and often include:- Fatigue and pallor, owing to anaemia and low haemoglobin;
- Easy bruising, petechiae (appearing as a purple rash, owing to bleeding under the skin), and unusual bleeding (which may be slow to stop owing to reduced platelet numbers in blood);
- Frequent infections owing to a low number of normal white blood cells[1].
Prognosis
The five-year survival rate for adults in the UK diagnosed with ALL is currently 35–40% and is particularly poor in older patients (<15% in patients aged over 65 years)[2]. Outcomes in children are much better, with >90% of patients aged 14 years or younger surviving for five years or more after diagnosis[2]. Prognosis in childhood ALL is related to five factors[5]:- Age at diagnosis — infants aged under one year are excluded from the standard paediatric protocols and receive a different, more intensified treatment protocol;
- Sex — trials showed that boys did less well than girls; therefore, boys now receive an extra year of maintenance chemotherapy;
- Presenting white cell count — children with a high white cell count have an inferior prognosis;
- Cytogenetics — non-random chromosomal alterations are seen in children at diagnosis. Some of these chromosome changes are associated with a good prognosis and some with an inferior prognosis. For example, Philadelphia chromosome-positive (Ph +) ALL has a poorer prognosis and is treated on a separate, more intensive treatment schedule;
- Response to treatment — bone marrow morphological examination and molecular quantification of residual leukaemia (MRD assessment) are used to assess response during and at the end of the first month of treatment[5],[6].
Overview of treatment
Treatment is risk-directed, meaning that children with a poorer prognosis receive more intensive treatment, while those with a good prognosis receive less intensive treatment. The aim is to not overtreat children who have a good prognosis and risk unnecessary treatment toxicity, but equally not to under-treat children who have an inferior prognosis and must not receive inadequate treatment. In general, protocols for children and young adults involve patients being assigned to a three- or four-drug remission induction schedule, known as schedules A and B, respectively[7],[8]. The schedule selected depends on the patient’s test results at diagnosis, age and white cell count. Patients who have a slow response to schedule A or B, or those who have other adverse features (e.g. high-risk cytogenetics) receive an intensified treatment, known as schedule C, after the first four weeks of treatment. In general, schedules A, B and C all include remission induction therapy (i.e. to kill the leukaemia cells in the bone marrow), consolidation therapy (i.e. treatment to stop ALL from recurring), interim maintenance therapy, delayed intensification and maintenance therapy (i.e. to keep ALL away in the long term). In children, the total treatment duration is just over two years for girls and three years for boys. The treatment of adult ALL follows a similar pathway, although the total duration of maintenance therapy is two years and is not sex-dependent. The medicines given during remission induction are as follows:- Vincristine — this is given intravenously, once per week for five weeks;
- Dexamethasone — this is a steroid with an antileukaemic action, given daily, usually orally;
- Pegaspargase — this is given by intramuscular injection in the first and the third week of treatment;
- Daunorubicin — this is given once a week via a central line during induction for schedule B patients or those switching to schedule C after the day-15 bone marrow test[9],[10],[11].
Other treatment strategies
Allogenic haematopoietic stem cell transplant (HSCT) is an alternative treatment option for patients:- Who do not respond to chemotherapy;
- With high-risk disease (e.g. Ph + ALL);
- With relapsed ALL.
New developments in treatment
There are two National Institute for Health and Care Excellence (NICE)-approved immunotherapy agents for the treatment of relapsed or refractory B-cell ALL[14],[15]. The choice of agent depends on which antigens are expressed by the leukaemia cell. Blinatumomab is a bi-specific T-cell engager antibody that targets the CD3 protein on T-cells and the protein CD19 on the tumour cell[16]. Whereas, inotuzumab ozogamicin is a conjugate of an anti-CD22 monoclonal antibody and a conventional cytotoxic agent[13]. NICE has also recently approved the use of blinatumomab for adult patients who achieve remission but have MRD activity[17],[18]. Data suggest that both drugs increase the time patients have without their disease relapsing and, therefore, the drugs may be used as a ‘bridge’ to HSCT or to the other significant new therapy, chimeric antigen receptor (CAR-T) therapy. In CAR-T therapy, the patient’s own T cells are removed and genetically modified so that they attach to and destroy leukaemia cells when they are given back to the patient by infusion. NICE has recently approved tisagenlecleucel, a form of CAR-T therapy that targets CD19, for the treatment of relapsed or refractory B-cell ALL in children and young adults aged up to 25 years[19]. Very encouraging clinical results have been reported with this novel approach, although it presents several logistical and economic challenges to the NHS[20]. For example, only selected centres in the UK are commissioned to deliver CAR-T therapy and, at the time of writing, there was not a formal way to record outcomes. These novel therapies may change the landscape of both childhood and adult ALL treatment in the next few years and there is much for pharmacists to consider in ensuring the safe delivery of these relatively new treatments to patients. For example, how to ensure that their governance is in-line with the safe and secure handling of medicines[21].Ongoing clinical trials
In the UK, a series of clinical trials have enabled the risk-directed approach to be taken forwards in the paediatric and young adult setting[7]. For example, ‘UKALL 2011’, a phase III randomised controlled trial, recruited children and young people between the ages of 1 and 25 years with ALL from April 2012 to December 2018[8]. The trial assessed whether changing the standard treatment for this patient group would reduce the side effects and help prevent the disease returning. Data analysis is currently ongoing. Furthermore, the current UK-wide national phase III randomised controlled trial in adult ALL (UKALL14) is investigating the benefits of adding novel agents (rituximab in B-cell ALL and nelarabine in T-cell ALL) to the standard induction chemotherapy schedule[13]. Patient recruitment ends in December 2020 and the trial is expected to end in 2022[13].Chronic lymphocytic leukaemia
Incidence
CLL is the most common leukaemia in adults in Western countries, with an age-adjusted UK incidence of 6 cases per 100,000 people per year[22]. It accounts for around 1% of all new cancer cases in the UK and the median age at diagnosis is 72 years[23]. The disease is more common in men, with a male to female ratio of 1.7:1 and there is evidence of a familial predisposition, with family members of patients with CLL having between a 6 to 9-fold increased risk of developing the disease[22],[23].Aetiology
The exact cause of CLL is unclear, but the increased familial risk of developing the disease suggests that an inherited genetic change may be involved[24].Presentation and diagnosis
In most cases, patients are asymptomatic and the disease is detected incidentally during a routine blood test. Symptoms, if present, may include lymphadenopathy, recurrent infections, fever, night sweats and weight loss. Cytopenias may also be seen, owing to infiltration of the bone marrow[25]. A diagnosis of CLL requires the presence of ≥5×109/L clonal B-lymphocytes in the peripheral blood for at least three months. Immunophenotyping, using flow cytometry, is necessary to confirm the clonality of the population. If the clonal population is smaller (i.e. <5×109/L) and the patient is asymptomatic, then the condition is designated as monoclonal B-lymphocytosis (MBL). This can be considered a pre-malignant condition and requires close follow-up because each year 1–2% of patients with MBL will go on to develop CLL[23]. Characteristic immunophenotypic features of CLL (e.g. high expression of CD5, CD23 and CD43, and low level expression of CD20 and CD79B) help to distinguish CLL from other conditions, such as mantle cell lymphoma, lymphoplasmocytic lymphoma and marginal zone lymphoma [23],[25]. CLL is staged by either the Binet or the Rai system; the former is more widely used in Europe (see Table 1), while the latter is preferred in the United States[26],[27]. Both systems divide patients into three groups based on physical symptoms and blood counts.| Table 1: Binet staging system for chronic lymphocytic leukaemia, using the five lymph node regions — cervical, axillary, inguinal, spleen and liver | ||
|---|---|---|
| Stage | Definition | Median survival |
| A | Haemoglobin >100g/LPlatelets >100×109/L Less than three impacted lymph node regions | >10.0 years |
| B | Haemoglobin >100g/LPlatelets >100×109/LThree or more impacted lymph node regions | >8.0 years |
| C | Haemoglobin <100g/L or platelets <100×109/L | 6.5 years |
| Adapted from Ann Oncol[23]; Blood[69] | ||
Prognosis
CLL is generally considered an incurable condition, although, as can be seen from Table 1, many patients will live for between five to ten years after diagnosis. As well as clinical stage, patient age and serum β-microglobulin concentrations have also been shown to influence prognosis[28]. As with many other malignancies, cytogenetic analysis (i.e. assessing an individual’s entire genome) is increasingly used to assist in predicting outcome and determining optimal treatment. A number of abnormalities (e.g. deletion 17p, deletion 11q and mutated TP53) are associated with a poorer outcome, while others (e.g. mutated immunoglobulin heavy chain variable) appear to confer a more favourable prognosis[29]. Around 5–10% of patients will be found to have TP53 disruption (either deletion 17p or mutated TP53) at diagnosis. Given that TP53 is a major tumour suppression gene, it is not surprising that these patients have been shown to have a poor prognosis and, as discussed in more detail below, usually require a different type of drug treatment to control their disease.Overview of treatment
Not all patients will require immediate treatment after receiving a diagnosis of CLL. Asymptomatic patients with early-stage disease are currently managed with a “watch and wait” approach because there is no evidence to support the use of chemotherapy in this subpopulation[30],[31]. However, recent data demonstrating a marked improvement in event-free survival with ibrutinib in asymptomatic patients who were determined to be at increased risk of progression, may lead to a re-evaluation of this position[32]. In 2018, the International Workshop on Chronic Lymphocytic Leukemia updated their treatment initiation guidance and their current criteria for starting treatment are summarised in the Box.Box: International Workshop on Chronic Lymphocytic Leukemia criteria for initiating chronic lymphocytic leukaemia treatment
Active disease should be clearly documented before therapy is initiated. At least one of the following criteria should be met:- Evidence of progressive marrow failure as manifested by the development, or worsening, of anaemia (haemoglobin <100g/L) and/or thrombocytopaenia (platelets <100×109/L);
- Splenomegaly that is massive (i.e. ≥6cm below the left costal margin), progressive or symptomatic;
- Lymphadenopathy with massive nodes (i.e. ≥10cm in longest diameter) or that is progressive or symptomatic;
- Progressive lymphocytosis with an increase of 50% or more over a two-month period, or a lymphocyte doubling time of less than six months. Factors contributing to lymphocytosis other than chronic lymphocytic leukaemia (e.g. infections or steroid administration) should be excluded;
- Autoimmune complications including anaemia or thrombocytopaenia poorly responsive to corticosteroids;
- Symptomatic or functional extranodal involvement (e.g. skin, kidney, lung or spine);
- Disease-related symptoms as defined by any of the following:
- Unintentional weight loss of 10% or more within the previous six months;
- Significant fatigue (i.e. Eastern Cooperative Oncology Group performance scale ≥2);
- Fevers (i.e. of 38.0°C or more) for two or more weeks without evidence of infection;
- Night sweats for one month or more without evidence of infection.
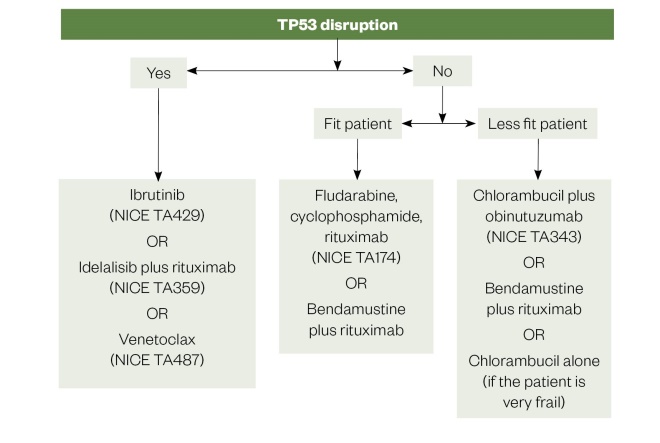
Figure 2: First line treatment options for chronic lymphocytic leukaemia
Source: National Institute for Health and Care Excellence; Br J Haem
NICE TA: National Institute for Health and Care Excellence technology appraisal
Ibrutinib
This oral agent targets Bruton tyrosine kinase (BTK), an important molecule in the B-cell receptor signalling pathway that drives cell proliferation[25]. It is taken on a continuous basis at a dose of 420mg once daily and has demonstrated impressive efficacy in both the first-line and relapsed CLL setting, and in patients with or without TP53 deletion or mutation[49],[50],[51]. For example, recently published extended follow-up data from the pivotal RESONATE study — which compared ibrutinib with ofatumumab in patients with relapsed CLL — demonstrated a 91% overall response rate for the ibrutinib arm and a significant benefit over ofatumumab in terms of both PFS and overall survival[52]. The tolerability profile of ibrutinib, as with other targeted therapies, is somewhat different to that seen with conventional chemotherapy[53]. Specific adverse effects to be aware of include diarrhoea, bleeding (inhibition of BTK has been shown to interfere with platelet aggregation), atrial fibrillation (three-fold increase in risk reported), hypertension, arthralgia/myalgia and an increased susceptibility to infection[54]. Marked lymphocytosis is often seen during the first few months of ibrutinib therapy, owing to tumour cells moving from the lymph nodes into the peripheral circulation. It is important that an increase in lymphocyte count is not taken as a sign of progressive disease but rather as an expected on-target effect of the ibrutinib[53]. Pharmacists need to be aware that ibrutinib is hepatically metabolised, primarily via CYP3A4, so it should be avoided, if possible, in patients who are taking strong CYP3A4 inducers (e.g. phenytoin, rifampicin) or inhibitors (e.g. clarithromycin, posaconazole)[53]. A careful medication review must be undertaken prior to starting ibrutinib, and patients should also be counselled about avoiding grapefruit and Seville oranges because they are predicted to increase exposure to ibrutinib[55].Idelalisib
This is an oral inhibitor of phosphatidylinositol -3-kinase, an intracellular tyrosine kinase that is also an important driver of B-cell proliferation via its effect on various signalling pathways. Idelalisib is given at a dose of 150mg twice daily, usually in combination with rituximab. Strong efficacy data in both the first-line and relapsed setting led to NICE approval[56],[57]. However, a subsequent analysis of clinical trial data demonstrated an increased rate of infection and death, therefore, its license was modified and it is now recommended that first-line use should be restricted to patients with TP53 disruption who are not eligible for any other therapy[58]. In light of this safety review, Pneumocystis jiroveci pneumonia (PJP) prophylaxis and regular monitoring for cytomegalovirus (CMV) infection are now recommended[59]. Concern has also been raised in relation to idelalisib- induced hepatotoxicity, possibly immune-mediated[60], and it is advised to monitor patients’ transaminases and total bilirubin every two weeks for the first three months of treatment[59]. Idelalisib is both metabolised by CYP3A and its primary metabolite is a strong CYP3A inhibitor so, as with ibrutinib, potential drug–drug and drug–food interactions need to be considered.Venetoclax
This is the most recent addition to the treatments available of CLL[61],[62]. It induces apoptosis by targeting the anti-apoptotic protein BCL2 that is known to be overexpressed in CLL cells. Venetoclax in combination with rituximab has recently been approved by NICE for use in the second-line setting, based on the results of the MURANO study of 389 patients that showed a marked difference in two-year PFS (85% vs. 36%, P<0.001) when compared with rituximab–bendamustine[63]. Venetoclax can also be used as a single agent in relapsed disease and in the first-line setting for patients with a TP53 deletion or mutation who are not suitable for treatment with a B-cell receptor pathway inhibitor[37],[38]. Common side effects of venetoclax include neutropenia, tumour lysis syndrome (TLS) and infection[29]. The risk of TLS is particularly high and a gradual dose escalation schedule (usually on an inpatient basis) must be followed for all patients starting treatment with this drug[40],[64] (see Table 2). Importantly, strong CYP3A4 inhibitors (e.g. clarithromycin and voriconazole) are contraindicated in patients receiving venetoclax. In patients who are receiving moderate CYP3A4 inhibitors (e.g. ciprofloxacin and fluconazole), dose adjustment of the venetoclax is mandated[64].| Table 2: Dose titration schedule for venetoclax | |
|---|---|
| Source: Electronic Medicines Compendium[64] | |
| Week | Venetoclax daily dose (mg) |
| 1 | 20 |
| 2 | 50 |
| 3 | 100 |
| 4 | 200 |
| 5 | 400 |
Other treatment strategies
Allogeneic stem cell transplant is a potentially curative option that may be considered for younger, fitter patients with relapsed or refractory CLL[25],[40]. Given the excellent results being seen with the novel targeted therapies in this disease, its future role is subject to ongoing debate[65]. However, it remains the standard of care for CLL patients with Richter’s transformation who have obtained a remission with conventional chemotherapy[40]. Maintenance therapy is increasingly used in other haematological malignancies, such as myeloma and acute myeloid leukaemia. Although PFS benefits have been demonstrated for anti-CD20 monoclonal antibodies and lenalidomide maintenance in the CLL setting, concerns about the long-term tolerability and a lack of an overall survival benefit mean that this strategy is not routinely recommended[25],[40],[66],[67],[68].Complications
In around 10% of patients with CLL, their disease will eventually transform to diffuse large B-cell lymphoma (DLBCL), a phenomenon called Richter’s transformation. Unfortunately, this usually has a poorer outcome than de novo DLBCL[23]. Transformation to Hodgkin lymphoma is also seen, albeit rarely. Healthcare professionals should be aware that patients with CLL have a two- to seven-fold increased risk of developing a secondary malignancy (mostly solid cancers)[23]. Therefore, patients should be encouraged to participate in national screening programmes (e.g. bowel cancer screening programmes). A reduction in humoral immunity is characteristic of CLL and this, taken alongside the myelosuppressive and immunosuppressive effect of treatment, means that patients are at increased risk of a variety of infectious complications. Most infections are bacterial but fungal, viral and opportunistic infections are also seen, necessitating careful monitoring and the use of prophylactic anti-infectives (e.g. co-trimoxazole and antivirals). Influenza and pneumococcal vaccinations are also recommended, and immunoglobulin replacement therapy should be considered in the setting of hypogammaglobulinaemia with recurrent infections[40]. Finally, autoimmune cytopaenias (most commonly autoimmune haemolytic anaemia or immune thrombocytopenic purpura) have been reported in up to 20% of patients with CLL.Conclusion
For both ALL and CLL, patient outcomes continue to improve, partly driven by improved clinical trial design and partly through the introduction of a range of novel treatments, focusing on molecular or immunological targets. However, further progress is required, particularly in the setting of adult ALL where many patients will die of their disease within five years of diagnosis.- This article was updated on 4 March 2019 to correct an error in Table 1. It previously stated “less than three impacted lymph node regions”; this should have read “Three or more impacted lymph node regions”.
References
[1] Children’s Cancer and Leukaemia Group. Acute lymphoblastic leukaemia (ALL). 2016. Available at: https://www.cclg.org.uk/publications/pgfactsheets/acute-lymphoblastic -leukaemia-ALL/PGFSALL (accessed January 2020)
[2] Cancer Research UK. Acute lymphoblastic leukaemia statistics. 2014. Available at: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/leukaemia-all (accessed January 2020)
[3] Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer 2018;18:471–484. doi: 10.1038/s41568 -018-0015-6
[4] Hoelzer D, Bassan R, Dombret H et al . Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2016;27(suppl 5):v69–v82. doi: 10.1093/annonc/mdw025
[5] Mitchell C, Richards S, Harrison CJ & Eden T. Long-term follow-up of the UK Medical Research Council protocols for childhood acute Lymphoblastic leukaemia, 1980–2001. Leukemia 2010;24:406–418. doi: 10.1038/leu.2009.256
[6] Pui CHI, Boyett JM, Relling MV et al . Sex differences in prognosis for children with acute lymphoblastic leukaemia. J Clin Onc 1999;17(3):818-824. doi: 10.1200/JCO.1999.17.3.818
[7] Vora A, Goulden N, Wade R et al . Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 2013;14(3):199–209. doi: 10.1016/S1470 -2045(12)70600-9
[8] ISRCTN clinical trial registry. UKALL 2011. 2019. Available at: http://www.isrctn.com/ISRCTN64515327 (accessed January 2020)
[9] BNF . Vincristine sulfate. 2018. Available at: https://bnf.nice.org.uk/drug/vincristine -sulfate.html (accessed January 2020)
[10] National Institute for Health and Care Excellence. Pegaspargase for treating acute lymphoblastic leukaemia. Technology appraisal guidance [TA408]. 2016. Available at: https://www.nice.org.uk/guidance/ta408 (accessed January 2020)
[11] BNF. Daunorubicin. 2017. Available at: https://bnf.nice.org.uk/drug/daunorubicin.html (accessed January 2020)
[12] Cancer Research UK. Acute lymphoblastic leukaemia. Chemotherapy drugs. 2018. Available at: https://www.cancerresearchuk.org/about-cancer/acute-lymphoblastic -leukaemia-all/treatment/chemotherapy/drugs (accessed January 2020)
[13] ISRCTN clinical trial registry. UKALL 14. 2019. Available at: http://www.isrctn.com/ISRCTN66541317 (accessed January 2020)
[14] National Institute for Health and Care Excellence. Blinatumumab for previously treated Philadelphia chromosome negative acute lymphoblastic leukaemia. Technology appraisal guidance [TA450]. 2017. Available at: https://www.nice.org.uk/guidance/ta450 (accessed January 2020)
[15] National Institute for Health and Care Excellence. Inotuzomab ozogamicin for treating relapsed or refractory B-cell acute lymphoblastic leukaemia. Technology appraisal guidance [TA541]. 2018. Available at: https://www.nice.org.uk/guidance/ta541 (accessed January 2020)
[16] Stackelberg A, Locatelli F, Zugmaier G et al . Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukaemia. J Clin Oncol 2016;34(36):4381–4389. doi: 10/1200/JCO.2016.67.3301
[17] Uly N, Nadeau M, Stahl M & Zeidan AM. Inotuzomab ozogamicin in the treatment of relapsed/refractory acute B cell lymphoblastic leukaemia. J Blood Medicine 2018;9:67–74. doi: 10.2147/JBM.S136575
[18] National Institute for Health and Care Excellence. Blinatumomab for treating acute lymphoblastic leukaemia in remission with minimal residual disease activity. Technology appraisal guidance [TA589]. 2019. Available at: http://www.nice.org.uk/guidance/ta589 (accessed January 2020)
[19] National Institute for Health and Care Excellence. Tisagenlecleucel for treating relapsed or refractory B-cell acute lymphoblastic leukaemia in people aged up to 25 years. Technology appraisal guidance [TA554]. 2018. Available at: https://www.nice.org.uk/guidance/ta554 (accessed January 2020)
[20] Maude SL, Laetsch TW, Buechner J et al . Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med 2018;378:439–448. doi: 10.1056/NEJMoa1709866
[21] Specialist Pharmacy Service. Pharmacy Institutional Readiness for Marketed CAR-T Therapy: Checklists for Pharmacy Services. 2018. Available at: www.sps.nhs.uk (accessed January 2020)
[22] Cancer Research UK. Chronic lymphocytic leukaemia statistics. 2019. Available at: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/leukaemia-cll (accessed January 2020)
[23] Eichhorst B, Robak T, Montserrat E et al . ESMO Guidelines Committee. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26 Suppl 5:v78–84. doi: 10.1093/annonc/mdv303
[24] Goldin LR, Björkholm M, Kristinsson SY et al . Elevated risk of chronic lymphocytic leukemia and other indolent non-Hodgkin’s lymphomas among relatives of patients with chronic lymphocytic leukemia. Haematologica 2009;94(5):647–653. doi: 10.3324/haematol.2008.003632
[25] Hallek M, Shanafelt TD & Eichhorst B. Chronic lymphocytic leukaemia. Lancet. 2018;391(10129):1524–1537. doi: 10.1016/S0140 -6736(18)30422-7
[26] Rai KR, Sawitsky A, Cronkite EP et al . Clinical staging of chronic lymphocytic leukemia. Blood 1975;46(2):219–234. PMID: 1139039
[27] Binet JL, Auquier A, Dighiero G et al . A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48(1):198–206. doi: 10.1002/1097-0142(19810701)48:1<198::aid-cncr2820480131 >3.0.co;2-v
[28] International CLL -IPI working group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL -IPI): a meta-analysis of individual patient data. Lancet Oncol 2016;17(6): 779–790. doi: 10.1016/S1470 -2045(16)30029-8
[29] Parikh SA. Chronic lymphocytic leukemia treatment algorithm 2018. Blood Cancer J 2018;8:93. doi: 10.1038/s41408 -018-0131-2
[30] Dighiero G, Maloum K, Desablens B et al . Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 1998;338:1506–1514. doi: 10.1056/NEJM199805213382104
[31] CLL Trialists Collaborative Group. Chemotherapeutic options in chronic lymphocytic leukemia. J Natl Cancer Inst 1999;91(10):861–868. doi: 10.1093/jnci/91.10.861
[32] Langerbeins P, Bahlo J, Rhein C et al . Ibrutinib versus placebo in patients with asymptomatic, treatment-naïve early stage chronic lymphocytic leukemia (CLL): primary endpoint results of the phase III double-blind randomized CLL12 trial. Presented at the European Hematology Association Congress, 13–16 June 2019; Amsterdam. Abstract LB2602.
[33] National Institute for Health and Care Excellence. Rituximab for the first line treatment of chronic lymphocytic leukaemia. Technology appraisal guidance [TA174]. 2009. Available at: https://www.nice.org.uk/guidance/ta174 (accessed January 2020)
[34] National Institute for Health and Care Excellence. Obinutuzumab in combination with chlorambucil for untreated chronic lymphocytic leukaemia. Technology appraisal guidance [TA343]. 2015. Available at: https://www.nice.org.uk/guidance/ta343 (accessed January 2020)
[35] National Institute for Health and Care Excellence. Idelalisib for treating chronic lymphocytic leukaemia. Technology appraisal guidance [TA359]. Available at: https://www.nice.org.uk/guidance/ta359 (accessed January 2020)
[36] National Institute for Health and Care Excellence. Ibrutinib for previously treated chronic lymphocytic leukaemia and untreated chronic lymphocytic leukaemia with 17p deletion or TP53 mutation. Technology appraisal guidance [TA 429]. Available at: https://www.nice.org.uk/guidance/TA429 (accessed January 2020)
[37] National Institute for Health and Care Excellence. Venetoclax for treating chronic lymphocytic leukaemia. Technology appraisal guidance [TA487]. 2017. Available at: https://www.nice.org.uk/guidance/TA487 (accessed January 2020)
[38] National Institute for Health and Care Excellence. Venetoclax with rituximab for previously treated chronic lymphocytic leukaemia. Technology appraisal guidance [TA561]. 2019. Available at: https://www.nice.org.uk/guidance/TA561 (accessed January 2020)
[39] National Institute for Health and Care Excellence. NICE Pathways: lymphoid leukaemia. 2019. Available at: https://pathways.nice.org.uk/pathways/blood-and-bone-marrow-cancers/lymphoid-leukaemia (accessed January 2020)
[40] Schuh A, Parry-Jones N, Appleby N et al . Guideline for the treatment of chronic lymphocytic leukaemia. A British Society for Haematology Guideline. Br J Haem 2018;182(3):344–359. doi: 10.1111/bjh.15460
[41] Hallek M, Fischer K, Fingerle -Rowson G et al . Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase III trial. Lancet 2010;376(9747):1164–1174. doi: 10.1016/S0140 -6736(10)61381-5
[42] Fischer K, Bahlo J, Fink AM et al . Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208–215. doi: 10.1182/blood-2015-06-651125
[43] Eichhorst B, Fink AM, Bahlo J et al . First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase III, non-inferiority trial. Lancet Oncol 2016;17(7):928–942. doi: 10.1016/S1470 -2045(16)30051-1
[44] Goede V, Fischer K, Busch R et al . Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014;370:1101–1110. doi: 10.1056/NEJMoa1313984
[45] National Comprehensive Cancer Network. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma guidelines 2019. Available at: https://www.nccn.org/professionals/physician_gls/pdf/cll.pdf (accessed January 2020)
[46] Moreno C, Greil R, Demirkan F et al . Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase III trial. Lancet Oncol 2019;20(1):43–56. doi: 10.1016/S1470 -2045(18)30788-5
[47] Jain N, Keating M, Thompson P et al . Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med 2019;380:2095–2103. doi: 10.1056/NEJMoa1900574
[48] Fischer K, Al-Sawaf O, Bahlo J et al . Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med 2019;380:2225–2236. doi: 10.1056/NEJMoa1815281
[49] Byrd JC, Brown JR, O’Brien S et al . RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med 2014;371:213–223. doi: 10.1056/NEJMoa1400376
[50] Burger JA, Tedeschi A, Barr PM et al . RESONATE-2 Investigators. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med 2015;373:2425–2437. doi: 10.1056/NEJMoa1509388
[51] O’Brien S, Furman RR, Coutre S et al . Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a five-year experience. Blood 2018;131(17):1910–1919. doi: 10.1182/blood-2017-10-810044
[52] Byrd JC, Hillmen P, O’Brien S et al. Long-term follow-up of the RESONATE phase III trial of ibrutinib vs ofatumumab. Blood. 2019;133(19):2031–2042. doi: 10.1182/blood-2018-08-870238
[53] Gribben JG, Bosch F, Cymbalista F et al . Optimising outcomes for patients with chronic lymphocytic leukaemia on ibrutinib therapy: European recommendations for clinical practice. Br J Haematol 2018;180(5):666–679. doi: 10.1111/bjh.15080
[54] Leong DP, Caron F, Hillis C et al . The risk of atrial fibrillation with ibrutinib use: a systematic review and meta-analysis. Blood 2016;128(1):138–140. doi: 10.1182/blood-2016-05-712828
[55] BNF . Ibrutinib. 2017. Available at: https://bnf.nice.org.uk/drug/ibrutinib.html (accessed January 2020)
[56] Furman RR, Sharman JP, Coutre SE et al . Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014;370:997–1007. doi: 10.1056/NEJMoa1315226
[57] O’Brien SM, Lamanna N, Kipps TJ et al . A phase II study of idelalisib plus rituximab in treatment-naïve older patients with chronic lymphocytic leukemia. Blood 2015;126(25):2686–2694. doi: 10.1182/blood-2015-03-630947
[58] Gilead Direct Healthcare Professional Communication. Restrictions on the use of Zydelig (idelalisib) for the treatment of chronic lymphocytic leukaemia (CLL) and relapsed follicular lymphoma (FL) following new clinical trial results. 2016. Available at: https://assets.publishing.service.gov.uk/media/5707baba40f0b60385000056/Zydelig __idelalisib __-_DHPC_sent_23_03_2016.pdf (accessed January 2020)
[59] Electronic Medicines Compendium. Zydelig 150mg tablets. 2019. Available at: https://www.medicines.org.uk/emc/product/3329/smpc (accessed January 2020)
[60] Lampson BL, Kasar SN, Matos TR et al . Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood 2016;128(2):195–203. doi: 10.1182/blood-2016-03-707133
[61] Stilgenbauer S, Eichhorst B, Schetelig J et al . Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase II study. Lancet Oncol . 2016;17(6):768–778. doi: 10.1016/S1470 -2045(16)30019-5
[62] Jones JA, Mato AR, Wierda WG et al . Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase II trial. Lancet Oncol 2018;19(1):65–75. doi: 10.1016/S1470 -2045(17)30909-9
[63] Seymour JF, Kipps TJ, Eichhorst B et al . Venetoclax -Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N Engl J Med 2018;378:1107–1120. doi: 10.1056/NEJMoa1713976
[64] Electronic Medicines Compendium. Venclyxto film coated tablets. 2019. Available at: https://www.medicines.org.uk/emc/product/2267 (accessed January 2020)
[65] Dreger P, Schetelig J, Andersen N et al . Managing high-risk CLL during transition to a new treatment era: stem cell transplantation or novel agents? Blood 2014;124(26):3841–3849. doi: 10.1182/blood-2014-07-586826
[66] van Oers MH, Kuliczkowski K, Smolej L et al . Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase III study. Lancet Oncol 2015;16(13):1370–1379. doi: 10.1016/S1470 -2045(15)00143-6
[67] Greil R, ObrtlÃková P, Smolej L et al . Rituximab maintenance versus observation alone in patients with chronic lymphocytic leukaemia who respond to first-line or second-line rituximab -containing chemoimmunotherapy: final results of the AGMT CLL -8a Mabtenance randomised trial. Lancet Haematol 2016;3(7):e317–329. doi: 10.1016/S2352 -3026(16)30045-X
[68] Fink AM, Bahlo J, Robrecht S et al . Lenalidomide maintenance after first-line therapy for high-risk chronic lymphocytic leukaemia (CLLM1): final results from a randomised, double-blind, phase 3 study. Lancet Haematol 2017;4(10):e475–e486. doi: 10.1016/S2352 -3026(17)30171-0
[69] Pflug N, Bahlo J, Shanafelt TD et al . Development of a comprehensive prognostic index for patients with chronic lymphocytic leukemia. Blood 2014;124(1):49–62. doi: 10.1182/blood-2014-02-556399
[70] Hallek M, Cheson BD, Catovsky D et al . iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018;131(25):2745–2760. doi: 10.1182/blood-2017-09-806398