Key points
- Immuno-oncology (IO) is emerging as a novel approach to cancer treatment through the stimulation of the body’s own immune system.
- Immune checkpoint inhibitors (ICPis) have had remarkable success across multiple malignancies, and are the most well-established IO agents to date, with several approvals.
- Biomarker testing for the programmed death-ligand 1 checkpoint target is obligatory before treating some tumour types with ICPis (e.g. pembrolizumab and atezolizumab).
- Combining IO agents with conventional therapies has provided significant improvements in patient outcomes in some cases.
- The two main challenges for IO agents are managing their toxicities and affording the high cost of these novel therapies.
Cancer learning ‘hub’
Pharmacists are playing an increasingly important role in supporting patients with cancer, working within multidisciplinary teams and improving outcomes. However, in a rapidly evolving field with numbers of new cancer medicines is increasing and the potential for adverse effects, it is now more important than ever for pharmacists to have a solid understanding of the principles of cancer biology, its diagnosis and approaches to treatment and prevention. This new collection of cancer content, brought to you in partnership with BeOne Medicines, provides access to educational resources that support professional development for improved patientIntroduction
Cancer incidence rates have steadily increased over the past 20 years, while mortality rates have shown a considerable decline[1] . Although significant variation in survival rates is still observed across cancer types (i.e. there are more 200 distinct diseases recognised), for most types, survival is improving owing to earlier diagnosis and improved treatments[2],[3] . Treatment has undergone a slow evolution from its start in the 1800s, with the sequential development of four main recognised modes of treatment. The first was surgery, which was made possible after the discovery of general anaesthetics in the late 1800s[4] . This was a revolutionary development because it was the first time the disease could be completely eradicated as long as the tumour was small and well-defined. The second development was radiotherapy, established at the end of the 19th century, which utilises X-rays and/or G-rays to damage the DNA within tumour cells, thus blocking essential biochemical processes and leading to cell death[4] . The third development, chemotherapy, was discovered in the 1940s, during World War II, when it was observed that individuals exposed to mustard gas suffered myelosuppression[4] . Clinicians speculated that patients with proliferative diseases (e.g. leukaemia) might benefit from treatment with agents of this type that kill highly proliferating cells. Crucially, introduction of the first chemotherapy agents (analogues of nitrogen mustard gas) meant that cancers which were more complex or had metastasised, and could not be successfully treated by surgery or radiotherapy, could now be addressed. Furthermore, chemotherapy agents have since been developed that work at different stages of the cell cycle, and can be used in combination to prevent the development of resistance. The fourth development was targeted cancer therapies (also known as precision therapies). This was established with the discovery of imatinib (Glivec; Novartis) in the late 1990s — a small-molecule kinase inhibitor targeted to the mutant BCR-ABL protein present in the tumour cells of patients with chronic myeloid leukaemia (CML), but not in their healthy cells[5] . This concept of using modern structural biology and drug discovery methods to produce small molecules, proteins, antibodies and even cellular therapies designed to target unique biomarkers associated with tumour cells, but not healthy cells, is now considered to be the ‘gold standard’ approach for discovering new cancer treatments. Currently, four major treatment modes — surgery, radiotherapy, chemotherapy and targeted agents — are frequently used in combination to ensure that all cancer cells are eradicated from the body. During the past decade, the first immuno-oncology (IO) treatments (e.g. checkpoint inihibitors) have emerged, which work by harnessing the body’s own immune system to kill tumour cells[6] . They are presently showing great promise in the clinic, and are the main focus of this review. Immune checkpoint proteins are found on the surface of T-cells and act as regulators of the immune system. They are crucial for self-tolerance, and prevent the immune system from attacking the body’s own cells indiscriminately, thus allowing a distinction to be made between ‘self’ and ‘non-self’[7] . Immune checkpoints also play a vital role in preventing uncontrolled immune responses by modulating the duration and amplitude of a physiological immune response, thus preventing collateral damage, which is why the term ‘off-switch’ is sometimes used to describe their role. It is known that tumours adopt certain immune checkpoint pathways as a mechanism to evade an immune response towards them[7] . For example, some tumour cell types express these proteins on their surface to disguise themselves as ‘self’, allowing them to go unnoticed by the immune system and promoting tumour progression[8] . PD-1 (programmed death 1) is an example of an inhibitory checkpoint receptor protein found on the surface of T-cells that normally acts as an ‘off-switch’ after interaction with the PD-1 ligand (PD-L1), a protein expressed on the surface of normal cells. However, PD-L1 is expressed by many types of tumour cells and upregulated in some, thus activating the ‘off-switch’ and protecting the malignant cells from an immune attack[9],[10] . Immune checkpoint inhibitors (ICPis), such as the anti-PD-1/PD-L1 agents, prevent the interaction between PD-L1 on tumour cells and PD-1 on T-cells, allowing the immune system to launch an antitumor response. Many observers believe that, over the next decade, IO agents could become the fifth acknowledged cancer treatment modality[11], [13] . Some of the main ligands and receptors present on the surface of tumour and immune cells that are targets for approved and emerging IO therapies are summarised in Figure 1.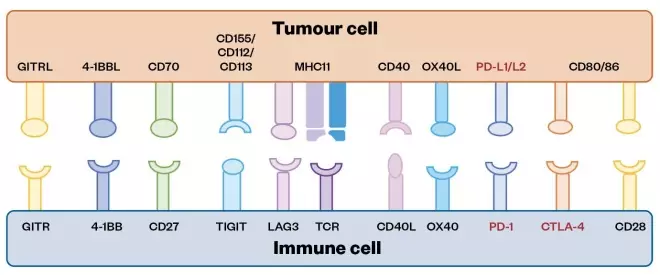
Figure 1: Some of the main ligands and receptors present on the surface of tumour and immune cells that are targets for approved and emerging immuno-oncology therapies
These targets are exploited by tumours to evade the immune system, and so represent an opportunity for immuno-oncology (IO) intervention. Ligands and receptors are represented as ball and cup structures, respectively, with approved therapies highlighted in red and emerging therapies highlighted in black. Other emerging IO targets are not located on the surface of cells, and so cannot be represented in this diagram (e.g. indoleamine 2,3-dioxygenase, which is an intracellular enzyme)
History of immuno-oncology
It has long been known, but is now increasingly appreciated, that tumour cells can be recognised and disabled by the immune system. Some tumours show evidence of spontaneous regression early in their development, suggesting that the immune system may be capable of recognising and eliminating early-stage tumour cells[14] . Observation of spontaneous remissions in patients led to the foundation of the area of IO. A spontaneous remission is defined as a reduction in severity of, or disappearance of, the signs and symptoms of a disease, without any apparent cause and in the absence of treatment. This is most often noted in patients who have recently had acute infections, especially when this results in fever which appears to stimulate the immune system. It is now recognised that, in some cases, the immune system is capable of completely eliminating a tumour. Spontaneous remissions have been observed in most cancer types, but most frequently in advanced melanoma, renal cell carcinoma (RCC) and urothelial carcinomas, although the phenomenon has also been reported in breast cancer, neuroblastomas, some sarcomas and embryonal cancers[15] . William Coley was the first to investigate the potential for IO, and successfully treated malignancies based on immune stimulation in the 1890s[16] . After discovering that cancer patients who contracted post-surgical infections seemed to improve faster than those who did not, he investigated the use of bacteria to stimulate and enhance the body’s natural immune response to fight cancer. Through these studies, he later developed Coley’s toxin, which was based on attenuated bacteria and is thought to be the first known IO therapy[17] . A later development involved the Bacillus Calmette-Guerin (BCG) vaccine, originally produced in the early 1900s for use against tuberculosis (TB), and first used therapeutically for TB in the 1920s. However, its role in cancer therapy dates back to 1929 when a reduced incidence of cancer among patients with TB was observed at autopsy[18] . Experiments revealed that BCG produced a profound stimulation of the mononuclear phagocyte system (also known as the reticuloendothelial system), which was recognised as an important defense against cancer. Furthermore, it was observed that neonates who had been immunised with BCG had a significantly lower incidence of leukaemia later in their lives[16] . This background and basic understanding of IO sparked an interest in the use of BCG for other types of malignancies, in particular bladder cancer. Early investigations demonstrated responses in patients with melanoma metastatic to the bladder when treated with intralesional BCG. In light of this success, work in animal models led to publication of the results of the first successful clinical trial of intravesical BCG in patients with recurrent bladder cancer[16] . It is now understood that intravesicularly adminstered BCG attaches to bladder tumours and urothelial cells via specific fibronectin and integrin receptors. Following internalisation by macropinocytosis, the mononuclear phagocyte system is stimulated by the BCG, inducing a local inflammatory response characterised by the infiltration of granulocytes, macrophages and lymphocytes. Important elements of the humoral immune response to BCG include the interleukins (ILs) IL-1, IL-2, IL-6, IL-8, IL-10, IL-12, tumour necrosis factor alpha (TNF-a) and interferon gamma (INF-g)[19] . More recently, studies have shown that BCG contains high levels of CpG oligodeoxynucleotide motifs that are known to induce the TNF-related apoptosis-inducing ligand (TRAIL) through IFN production[16] . Intravesical BCG is still indicated for the treatment and prevention of recurrence of some types of non-invasive bladder cancers[20],[21] .Classification ofimmuno-oncology agents
The categorisation of IO agents is challenging and there is significant crossover and ambiguity with emerging agents. The classification devised and utilised throughout this review is represented in Tables 1–4 (a full PDF of the article, including all tables, can be found here). For example, ICPis (see Table 1) are sometimes classified separately to monoclonal antibodies (mAbs) (see Table 2), yet the ICPis are, themselves, monoclonal antibodies. The Cancer Research Institute takes two broad approaches to classification based on treatment type or cancer type[22] . Few observers employ the three very broad categories that have emerged over the years: non-specific cytokines, cancer vaccines and mAbs[23] .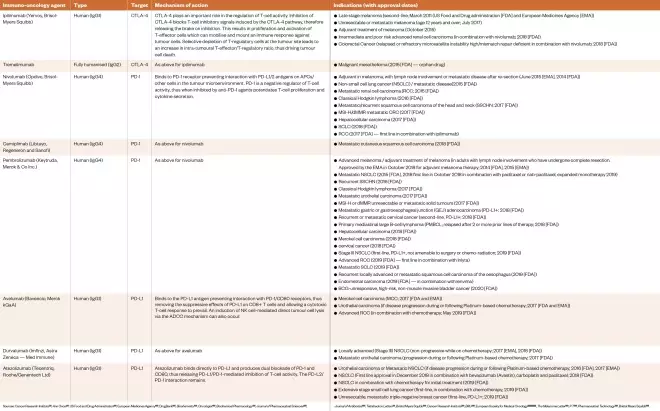
Table 1: Approved immune checkpoint inhibitors as of April 2020
Sources: Cancer Research Institute[22] , Ann Oncol [27] , US Food and Drug Administration[133] , European Medicines Agency[134] , DrugBank[135] , Biochemistry [136] , Oncologist [137] , Biochemical Pharmacology [138] , Journal of Pharmaceutical Sciences [139] , Journal of Antibiotics [140] , Tetrahedron Letters[141] , Bristol Myers Squibb[142] , Cancer Research Institute[143] , BMJ [144] , European Society for Medical Oncology[145] ,[147] ,[151] , The Melanoma Letter [146] , P T [148] , Pharmaceutical Technology[149] , Bristol Myers Squibb[150]
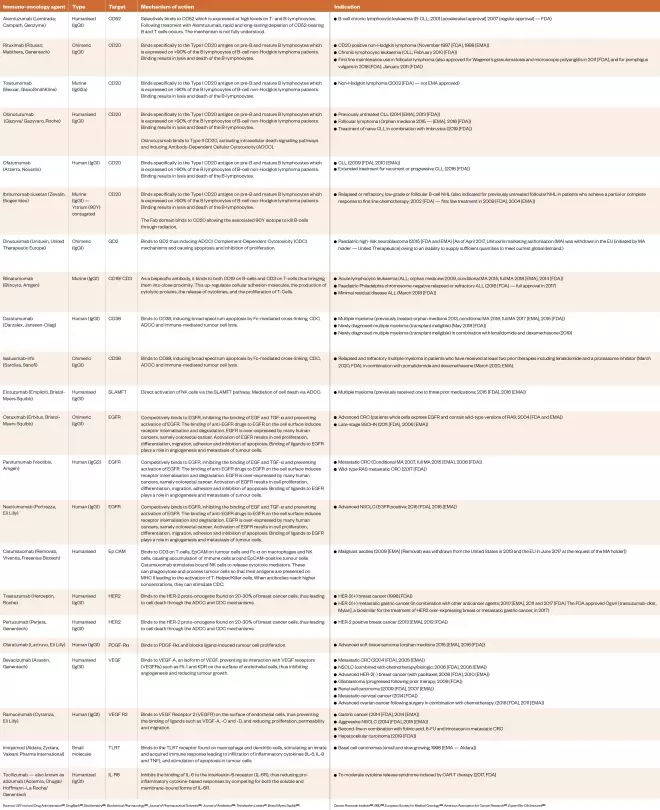
Table 2: Approved immuno-oncology agents directed against external and internal targets as of April 2020
Sources: US Food and Drug Administration[133] , DrugBank[135] , Biochemistry [136] , Biochemical Pharmacology [138] , Journal of Pharmaceutical Sciences [139] , Journal of Antibiotics [140] , Tetrahedron Letters [141] , Bristol Myers Squibb[142] , Cancer Research Institute[143] , BMJ [144] , European Society for Medical Oncology[145] , American Association for Cancer Research[152] , Expert Rev Clin Immunol[175]
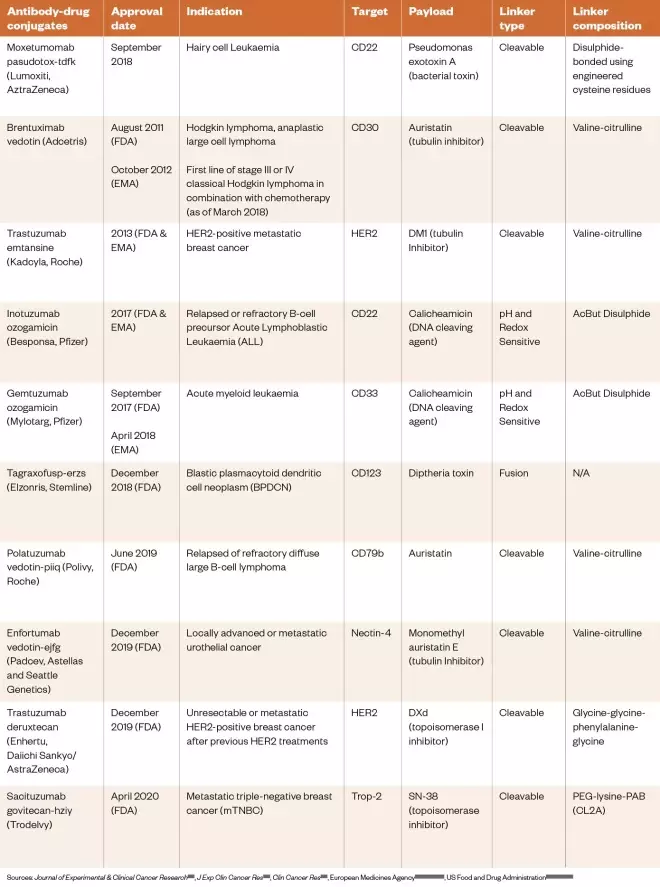
Table 3: Approved antibody-drug conjugates and immunotoxins as of April 2020
Sources: Journal of Experimental & Clinical Cancer Research [153] , J Exp Clin Cancer Res [154] , Clin Cancer Res [155] , European Medicines Agency[156] ,[159] ,[160] ,[162] , US Food and Drug Administration[157] ,[158] ,[161] ,[163]
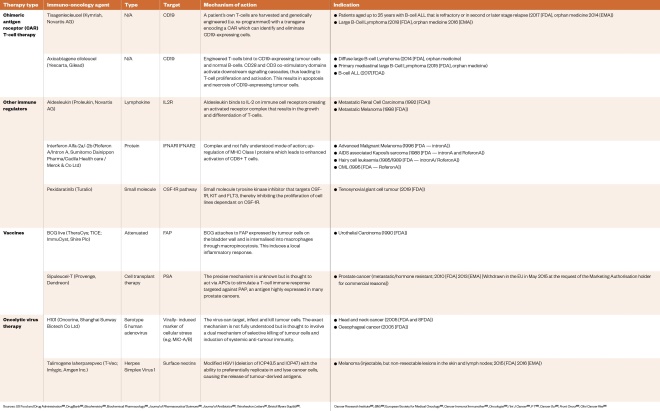
Table 4: Approved immuno-oncology therapies with an ‘active’ mechanism of action as of April 2020
Sources: US Food and Drug Administration[133] , DrugBank[135] , Biochemistry [136] , Biochemical Pharmacology [138] , Journal of Pharmaceutical Sciences [139] , Journal of Antibiotics [140] , Tetrahedron Letters [141] , Bristol Myers Squibb[142] , Cancer Research Institute[143] , BMJ [144] , European Society for Medical Oncology[145] , Cancer Immunol Immunother [164] , Oncologist [165] , I Int J Cancer [166] , P T [167] , Cancer Sci [168] , Front Oncol [169] , Clini Cancer Res [170]
Pharmacogenomic and precision medicine approaches to immuno-oncology
Drug discovery and development in the IO area is moving rapidly toward a pharmacogenomic approach, where biomarkers are identified in biopsy material from tumours so that predictions can be made about which therapies would be the most efficacious for a given patient[28],[29] . A recent retrospective study in a large number of pancreatic cancer patients (1,856) highlighted the significant effect a precision medicine approach can have on survival, particularly in cancer types with poor outcomes[172] . In this study it was shown that patients with actionable mutations (including some associated with checkpoint inhibitors) who received matching targeted therapies had longer overall survival times by up to 1.07 years compared to those receiving only unmatched therapies, respectively. For the main families of IO agents, the anti-PD-1/PD-L1 and anti-cytotoxic T-lymphocyte-associated protein-4 (anti-CTLA-4) agents, the clinical data relating to target expression and response to therapy is complex. For example, there are reports of responses to treatment irrespective of PD-L1 expression. There is also ambiguity around the thresholds used to define ‘positive’ and ‘negative’ biomarker expression values. For example, for PD-L1, ‘weak positive’ is defined as 1–49% expression and ‘strong positive’ as greater than 50% expression[30] . These broad definitions suggest that PD-L1 is not a clear dichotomous biomarker, and there is a need to find new biomarkers for IO treatments with increased specificity and reproducibility. The PD-1/PD-L1 biomarker assays available, the response rates in PD-L-positive and PD-L-negative patients, and emergent biomarkers are briefly addressed below.PD-1/PD-L1 biomarker assays
The PD-L1 ligand, which is expressed on the surface of some tumour cell types, is a vital molecular target for around half of all ICPis approved to date. Binding of this ligand to PD-1 receptors, expressed on the surface of T-cells, blocks their inhibitory activity toward tumour cells. PD-L1 is also expressed by various normal cells but is up-regulated in tumour cells and tumour-infiltrating immune cells, thus protecting them from an immune response[31] . Therefore, testing patients for tumour cell PD-L1 expression may lead to better clinical outcomes if they are selected for treatment with anti-PD-L1 agents. Early clinical studies investigating PD-L1 expression and the subsequent response of patients to the anti-PD-L1 agent nivolumab (Opdivo, Bristol-Myers Squibb) demonstrated the potential benefit of pharmacogenomic testing; in PD-L1-positive patients the objective response rate was 36%, while in PD-L1-negative patients there were no responses[32] . However, later reports from other clinical trials (e.g. NCT01642004, NCT01668784 and NCT02008227) showed that positive responses with prolonged overall survival can occur (compared with current standards of care) in PD-L1-negative patients[33],[34],[35],[36] . Therefore, based on the results of meta-analyses of clinical trial data, it is evident that PD-L1 expression status alone is insufficient to determine whether patients should be offered PD-1 or PD-L1 therapy[33] . Nivolumab (anti-PD-1) and atezolizumab (anti-PD-L1) were both approved without PD-L1 testing, while pembrolizumab (anti-PD-L1) is approved for first-line treatment of non small-cell lung cancer (NSCLC) only after testing patients for PD-L1 expression. An immunohistochemistry (IHC) test is used to determine biomarker expression, with a threshold set for first-line clinical use of the agent. PD-L1 expression must be greater than 50% using the Dako 22C3 IHC assay, whereas for second-line treatment only greater than 1% expression is required. However, another study has shown that patients with 5% or more positivity do not have a benefit over standard chemotherapy[28] . Complementary PD-L1 tests have been approved by the US Food and Drug Administration (FDA), but are not mandatory other than for pembrolizumab (which is a companion test; see Table 5)[37] . There are currently four other companion PD-L1 assays in development for PD-1/PD-L1 inhibitors[28] .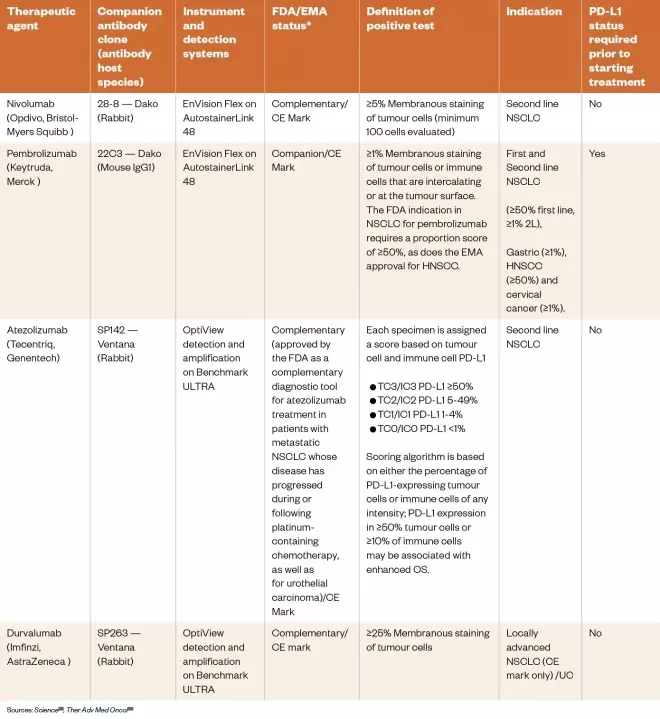
Table 5: Immunohistochemistry assays to measure PD-L1 expression
Response rates in PD-L1-positive and PD-L1-negative patients
There are multiple reports of PD-L1-negative patients responding to anti-PD-L1 IO agents, and immunostaining of tumour biopsies has revealed a possible explanation for this[40],[41] . There can be significant heterogeneity of expression of PD-L1 across an individual biopsy, with areas of no or very low PD-L1 expression but others with very dense expression[42] . Therefore, a patient may be categorised as PD-L1-negative if the area analysed from a biopsy shows no staining, whereas other regions of the tumour missed during the biopsy may have dense PD-L1 expression. Thus, it may not be possible to conclude from a single biopsy whether a patient is definitively PD-L1-negative or PD-L1-positive. While a higher level of PD-L1 expression has been associated with more favourable response rates to anti-PD-1/PD-L1 agents in some studies, positive responses have also been observed in some PD-L1-negative patients. Therefore, PD-L1 does not appear to offer binary discrimination of responsiveness[43] . Another possible contributory factor is that PD-L1 is not a static biomarker, but is dynamic with the degree of expression dependant on many biological processes. For example, there are genetic-based mechanisms that lead to constitutive PD-L1 expression, although expression can also be induced by the presence of T-cells[44] . Therefore, a tumour may be PD-L1-negative at a given point in time because there is no T-cell infiltrate, but this situation may be reversed owing to an immune response that itself may be stimulated by treatment with IO agents. Finally, biomarker heterogeneity of expression can arise owing to a variety of other factors, including: the stage of disease; prior treatments (e.g. type of chemoradiotherapy); tumour mutation status (e.g. PD-L1 expression in NSCLC is regulated by several oncogenic drivers, such as estimated glomerular filtration rate and anaplastic lymphoma kinase, that can alter expression levels); and concomitant medication use (e.g. corticosteroids)[45] .Emergent biomarkers
There are too many emergent biomarkers in the IO area to describe in detail in this review; however, some examples are outlined below. Tumour mutational burden (TMB) is a measure of the number of mutations within a tumour genome, and a high TMB has been shown to be associated with a favourable outcome for ICPis. For example, many tumours that respond to anti-PD-1 agents (e.g. melanoma, NSCLC and bladder cancer) have a high mutational load[28] . Some studies have attempted to correlate mutational load in NSCLC and melanoma with a response to ICPis, but the results have been unable to prove that a high mutational load alone enhances the response to therapy, therefore, its clinical utility is presently unclear[46] . Although clinically validated biomarkers for predicting response to pembrolizumab include PD-L1 expression (in specific tumour types) and high microsatellite instability (MSI-H; independent of tumour type), several emergent IO-related biomarkers associated with improved overall response rate (ORR) and progression-free survival (PFS) for ICPis are being studied. These include T-cell-inflamed gene expression profile (GEP), TMB and mutated mismatch repair (MMR) genes. PD-L1 and GEP are both inflammatory biomarkers associated with a T-cell-inflamed tumour microenvironment, whereas MSI-H and TMB are indirect measures of tumour antigenicity derived from somatic tumour mutations. In a 2018 study of more than 300 advanced solid tumour and melanoma samples from across 22 cancer types from four KEYNOTE clinical trials, Cristescu et al. assessed the potential for TMB and T-cell-inflamed GEP to jointly predict clinical response to pembrolizumab, with patients stratified into four biomarker-defined clinical groups of: GEP low/TMB low; GEP low/TMB high; GEP high/TMB low; and GEP high/TMB high[28] . The analysis showed that TMB and inflammatory biomarkers (i.e. T-cell-inflamed GEP and PD-L1 expression) can jointly stratify human cancers into groups with different clinical responses to pembrolizumab monotherapy, and that TMB and inflammatory biomarkers independently predict response and may be associated with neoantigenicity (the formation of new antigens not previously seen by the immune system) and T-cell activation, respectively. Overall, these studies found that longer PFS was observed for patients with high TMB and GEP values with a modest correlation between the two, although TMB and GEP could also predict response independently. However, determining TMB in tissue samples has several limitations, including heterogenous sample characteristics and a dependence on the timing of the assay. Furthermore, assays used to evaluate TMB have not been standardised, and the definition of “high” TMB varies significantly across different studies[47] . Most clinical studies carried out to date have been based on a variety of techniques making it difficult to compare available data and collate sufficient evidence to support its clinical use[48] . Finally, it has long been established that loss of function mutations in the MMR pathway are associated with favourable responses towards PD-1 blockade therapy, hence the interest in using MMR as a biomarker to predict responses. In an expansion of a proof-of-concept study reported in 2017 investigating disease progression in patients with MMR deficiencies across 12 different tumour types, all patients had been previously treated with pembrolizumab for anything up to 2 years[49] . Positive results were noted across all tumour types, with 77% of patients attaining disease control for at least 12 weeks, including 18 who had complete responses. Therefore, MMR deficiency is now considered a viable biomarker for patient selection for treatment with pembrolizumab[49] . CD45RA is an example of an emerging biomarker for anti-CTLA-4 agents. Its baseline expression level in T-cells has been found to correlate with clinical response to anti-CTLA-4 agents. Patients with higher numbers of CD45RA- cells compared to CD45RA+ cells in both CD4 and CD8 T-cell compartments responded more effectively to anti-CTLA-4 treatment. The CD45RA- biomarker is associated with induction of central and effector memory T-cells, and so these results suggest that the CD45RA status of baseline memory CD4 and CD8 T-cells and CD8 effector memory T-cells may be used to predict response to anti-CTLA-4 treatment[50] . Another study looking at clinical response in melanoma patients treated with ipilimumab found that patients with normal baseline levels of CD45RO+ CD8 T-cells responded more frequently to treatment, and a significantly longer overall survival (OS) was also observed in normal baseline CD45RO+ patients[51] . As IO agents are associated with potentially life-threatening toxicities, some observers have suggested that it would be preferable for the focus of biomarker research to shift toward attempting to predict toxicity rather than therapeutic response, thus identifying patients who may better tolerate treatment and gain overall benefit. This approach may be particularly important for combination therapies, which are generally associated with a greater frequency of high-grade toxicities.Increasing the responsiveness of tumours to the immune system
Tumour immunogenicity varies significantly between cancers of the same type in different individuals, and between different malignancies. Some cancers, such as pancreatic ductal adenocarcinoma are considered to be non-immunogenic (i.e. lacking the ability to induce an immune response)[52] . Common features of non-immunogenic tumours include a lack of tumour-infiltrating lymphocytes (TILs) and a lower response to immunotherapy[52],[53] . An elevated neutrophil/lymphocyte ratio (from baseline) has been correlated with poor patient outcomes following immunotherapy across multiple cancer types[54] . A promising related treatment strategy has emerged based on categorising tumours as ‘hot’ or ‘cold’ from an immunological perspective. For example, if the tumour microenvironment contains antigen-specific CD8 TILs it is regarded as ‘hot’, because the extent of infiltration of lymphocytes correlates with the degree of inflammation, and this has the potential to act as a biomarker to determine whether a tumour will respond to IO therapy. The aim is to transform ‘cold’ tumours into ‘hot’ tumours, thus increasing their responsiveness to IO agents and to prevent these tumours from ‘cooling off’ and becoming unresponsive to therapy. Another proposed method to increase the immunogenicity of tumours is to administer an oncolytic virus to promote a strong anti-viral immune response[55] . The resulting cytokine production (e.g. type-1 interferons) can directly promote the expression of PD-L1, while chemokines (e.g. CCL3 and CCL4) can attract PD-1/CTLA-4-expressing immune cells. This increased expression of cell-surface targets and infiltration of immune cells can facilitate the binding of ICPis and facilitate their effects. A small phase Ib trial has demonstrated that intralesional injection of herpes simplex virus (i.e. talimogene laherparepvec) in combination with systemic anti-PD-1 treatment results in a 62% ORR (and 33% complete response rate) in patients with metastatic melanoma. This treatment strategy is accompanied by an increased TIL presence, which has been interpreted as an alteration of the tumour micro-environment from immunologically ‘cold’ to inflamed and tumour responsive (i.e. ‘warm’)[56] . Although chemotherapy and radiotherapy are generally regarded as immunosuppressive, it is now accepted that they can work synergistically with IO-based therapies to achieve additive clinical benefit[57] . The mechanism is thought to involve induction of immunogenic cell death by chemotherapy that causes the release of damage-associated molecular patterns — host biomolecules with the ability to initiate an inflammatory immune response that can increase the responsiveness to IO agents. Radiation-induced cancer cell damage can expose tumour-specific antigens, thus making them visible to the immune system and leading to promotion of the priming and activation of T-cells[58] . Radiotherapy can also modulate the tumour microenvironment to facilitate the infiltration of immune cells, and can activate innate and adaptive immune responses through the stimulation of STING (stimulator of interferon genes), a pathway that plays a critical role in anticancer immunity.Evidence of efficacy
IO agents focus on the tumour micro-environment, thus allowing the immune system to produce efficient antitumour responses via negative regulatory pathways such as PD-1/PD-L1 and CTLA-4[59] . The ICPis (see Table 1) have consistently provided outstanding clinical outcomes across many tumour types (e.g. NSCLC and advanced RCC), leading to many accelerated approvals from the FDA and European Medicines Agency (EMA), regardless of PD-L1 and CTLA-4 expression status. Approvals and clinical guidance are based upon three main outcome measures: OS, PFS and ORR. The KEYNOTE-407 trial found statistically significant improvements in OS, PFS and ORR for patients receiving pembrolizumab plus chemotherapy, compared with randomised placebo plus chemotherapy, in patients with NSCLC, regardless of histologic subtype or PD-L1 expression. The investigators concluded that there was a high level of activity coÂÂmpared with chemotherapy alone for PD-L1-negative patients[60], [61] . Significant PFS and OS were also observed across differing PD-L1 expression levels in patients with NSCLC who had received durvalumab (Imfinzi, AstraZeneca) following chemoradiation treatment[62] . Durvalumab has also shown significant activity in late-stage NSCLC; study results showed that it significantly increased the OS rate at 24 months to 66.3% compared with 55.6% in the placebo groups, and PFS was also improved by more than 30% when compared with placebo (i.e. 17.2 months vs. 5.6 months, respectively). This result was pivotal because it was the first time that an IO treatment had improved survival in patients with late-stage NSCLCÂÂÂ[63] . In addition, the findings of a phase III randomised trial evaluating the use of an IO–biologic combination therapy, compared with a biologic monotherapy in patients with advanced RCC, revealed PFS and ORR benefits in patients irrespective of PD-L1 expression[40] . In addition, a randomised controlled study revealed that nivolumab was associated with higher ORRs than chemotherapy in patients with ipilimumab-resistant metastatic melanoma. The ORR for nivolumab was 40.0% — significantly higher than that for dacarbazine at 13.9%, and complete response rates were 7.6% and 1.0%, respectively. The subgroup analyses found that nivolumab-treated patients had improved OS when compared with chemotherapy, regardless of PD-L1 status[64] . While these studies implied that determining PD-L1 expression prior to treatment was of little importance, other reports suggested improved outcomes for PD-L1-positive patients[65] . Researchers who studied the effects of adding nab-paclitaxel to atezolizumab therapy in triple-negative breast cancer patients emphasised the importance of determining PD-L1 expression status to inform treatment choices, with their results suggesting that most of the benefit, but not all, was realised in the PD-L1-positive subgroup[66] . Although the current approvals are for combinations with existing therapies, the likely growth of further approvals of both single and combination therapies in the near future could alter the standard of care across many tumour types in the next decade and beyond. The PD-1/PD-L1 pathway is now considered to be one of the most promising areas of IO, and is the backbone of IO research and development[67] . For example, all six approved anti-PD-1/PD-L1 agents received approval within a three-year period (see Table 1), and clinical trial data have demonstrated activity across many different tumour types with prolonged and durable responses. Although PD-1/PD-L1 modulation is considered to be a relatively established area of IO, there are ongoing efforts to discover novel agents of this class to treat new indications. Importantly, five-year follow-up data from the phase 3 KEYNOTE-006 trial reported in 2019 have confirmed that pembrolizumab is superior to ipilimumab for the treatment of melanoma in patients who have had no more than one prior systemic therapy[68] . The median OS was 32.7 months for pembrolizumab versus 15.9 months for ipilimumab. There are currently only two anti-CTLA-4 agents (ipilimumab and tremelimumab) approved for use in both melanoma and mesothelioma. Although this narrow spectrum of indications compared with the anti-PD-1/PD-L1 agents suggests this might be an area for further development, there have been multiple reports of high-grade toxicities[69] . Despite this, CTLA-4 blockade is associated with durable and consistent survival benefits in some patients[70] . Therefore, researchers are keen to find ways to manage or overcome these toxicities so that new indications and combination therapies can be explored.Current research
As of September 2017, 58% of all clinical trials evaluating IO therapies were combination trials, 82% of which involved either another IO agent, a targeted therapy and/or a cytotoxic agent, while around 16% of combination trials involved PD-L1 antagonists and 20% CTLA-4 inhibitors[70] . However, as of September 2019, there were 1,469 more active clinical trials evaluating PD-1/PD-L1 mAbs alone or in combination with other agents, with 76% of these active trials investigating combination therapies[71] . NSCLC, melanoma and non-Hodgkin’s lymphoma have been at the forefront of IO research since its infancy, although, in recent years, interest in other malignancies such as renal, pancreatic and advanced (metastatic) cancer have significantly increased[72] . However, since 2014 the average number of planned enrolments has declined from an average of 429 to 129 patients per trial, reflecting the shift in focus from major tumour types (e.g. melanoma and breast cancer) to rarer cancers with a significantly smaller eligible population[71] . Current clinical research efforts are focussed largely on combining recently approved IO agents with either another IO agent or an existing treatment (i.e. chemotherapy or radiotherapy). Data from 2018 show that there are more than 1,700 clinical trials worldwide assessing combinations of anti-PD-1/PD-L1 agents with other cancer therapies, including anti-CTLA-4 agents (n=339), chemotherapy (n=283) and radiotherapy (n=114)[72] . This shift from monotherapies to combination therapies within clinical trials has resulted in 14 approvals of combination therapies by the FDA, with the three most common being PD-1/PD-L1 inhibitors in combination with chemotherapy, CTLA-4 inhibitors and vascular endothelial growth factor (VEGF)-targeted therapies (as of September 2019)[71] . T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domains (TIGIT) is an immune receptor present on the surface of some T-cells and natural killer (NK) cells. Similar to PD-1, it is an inhibitory checkpoint that is upregulated in multiple cancer types (e.g. melanoma, colon and renal cancer) and also plays a role in the activation and maturation of T-cells and NK cells[73] . The associated ligand, poliovirus receptor (PVR), is highly expressed on the surface of dendritic, endothelial and some tumour cells. TIGIT plays a vital role in suppressing the antitumour immune response within the tumour microenvironment. Therefore, blockade of binding to the ligand PVR may suppress its immunosuppressive signalling and allow the co-receptor CD-226 pathway to resume its T-cell activating functions[74] . The NCT02794571 and NCT02913313 trials are investigating TIGIT-blocking antibodies, both as monotherapy or as part of a combination with the PD-1/PD-L1 blocking antibodies atezolizumab and nivolumab, respectively[75] . The trial NCT03119428 that was evaluating the safety and tolerability of OMP-31M32 as a single agent or in combination with nivolumab was terminated in 2019[76] . OX40, a member of the TNF receptor family, is primarily expressed on CD8 T-cells, NK cells and neutrophils. Its ligand, OX40L, is expressed by B-cells and macrophages, and binding of OX40 to its ligand modulates T-cell activation and effector function. Studies in pre-clinical models have demonstrated that anti-OX40 antibodies can increase antitumour immunity and improve tumour-free survival[77] . Currently, multiple OX40-targeted antibodies are being evaluated in several phase I/II clinical trials, either as a monotherapy or in combination with other IO agents[74],[75],[76],[77],[78],[79] . Recent evidence suggests that activation of the STING pathway, a major innate immune pathway, is involved in the generation of spontaneous antitumour T-cell responses. STING activation within antigen-presenting cells in the tumour microenvironment leads to production of IFNb and spontaneous generation of antitumour CD8 T-cell responses. In addition, it has been observed that a deficiency in this pathway increases susceptibility to tumour progression. Therefore, the deliberate activation of the STING pathway has been identified as a major research area for the future[80] . Indoleamine-pyrrole 2,3-dioxygenase (IDO) is a heme-containing enzyme encoded by the IDO1 gene. With other related enzymes it catalyzes the first and rate-limiting step in the kynurenine pathway (i.e. the oxygen-dependent oxidation of L-tryptophan to N-formylkynurenine). It has been implicated in immune modulation by limiting T-cell function and engaging mechanisms of immune tolerance. Previous studies have suggested that IDO becomes activated during tumour development, helping malignant cells escape eradication by the immune system. Furthermore, IDO expression is closely linked to both CTLA-4 and PD-1/PD-L1 expression via several complex pathways. For example, the IDO enzyme, which is an intracellular target, can be induced by the interaction of PD-1 with PD-L1 on the surface of mast cells. It is expressed by various tumour types and, in many, high IDO expression correlates with poor survival and prognosis[81] . Pre-clinical and early phase clinical trials have shown that a combination of CTLA-4 blockade with IDO inhibition can provide more effective antitumour immunity, making IDO a potential novel target for IO therapy. Current IDO inhibitors are small-molecule rather than antibody-based, and examples include indoximod, epacadostat and navoximod, which have been studied both alone and in combination. Another experimental IO target is the glucocorticoid-induced tumour necrosis factor receptor (GITR), a surface receptor molecule involved in inhibiting the suppressive activity of T-regulatory cells and extending the survival of T-effector cells. Thus, GITR has the capacity to promote effector T-cell functions and impede T-regulator suppression. The anti-GITR antibody TRX518 was developed to target GITR and bind in an agonistic fashion. This agent reached phase I clinical trials in 2010, with safety reports published in 2019, and led to combination studies with anti-PD-1 agents in patients with advanced refractory tumours[82] . Another study investigated the use of the anti-GITR antibody MK-4166, both as a monotherapy and in combination with pembrolizumab[82] . Overall the results showed that mild immune-related adverse effects(irAEs) were common, occurring in more than 20% of patients after treatment with MK-4166 in combination with pembrolizumab, with only one dose-limiting toxicity of bladder perforation in a urothelial patient-reported. An ORR of 69% was achieved in ICPi-naïve melanoma patients, which included four complete and five partial responses. However, although these results were promising, the sample size was small (n=13). Lymphocyte activation gene 3 (LAG3), also known as CD223, is a cell surface protein expressed on activated CD8+ T-cells and other immune cells, which enhances the regulatory T-cell activity and negatively regulates cellular proliferation and activation and T-cell homeostasis. It specifically inhibits CD8+ effector T-cell functions and can enhance the suppressive activity of T-regulators. Multiple models have demonstrated that blockade of LAG3 with mAbs can augment T-cell function, although the mechanism(s) by which this occurs are poorly understood[83] . LAG3 is often co-expressed with other inhibitory proteins, especially PD-1. Several pre-clinical studies have suggested the potentially greater therapeutic benefit of dual blockade of these receptors (LAG3 and PD-1) compared with a single agent blockade. The dual blockade approach has demonstrated promising survival benefits and durable response rates in early phase I clinical trials in small subgroups of patients with specific cancer types (e.g. RCC), although detailed knowledge of the biology of LAG3 is presently lacking[83] . There is also significant ongoing research in the pre-clinical area, with a notable increase in efforts to identify and evaluate new IO drug targets. For example, in 2017, 165 targets were being evaluated, while in 2018 this increased by around 45% to 240 targets[72] .Toxicity
Owing to their mechanism of action, IO agents are associated with a unique but variable spectrum of toxicities, known as irAEs[84],[85] . While the toxicities can vary greatly depending on the patient, the risk of serious clinical problems developing limits the use of IO agents to specialist clinicians with the experience to deal with irAEs should they arise. The most serious concern is potential supra-physiologic stimulation of the immune system leading to a potentially life-threatening uncontrolled and rapid production of pro-inflammatory cytokines (a so-called ‘cytokine storm’) [85]. Since there are several potential irAEs that can present following initiation of IO therapy, it is important to have clear and robust guidelines of when to refer to other medical specialists who are able to provide input to managing individual patients. For this to work in practice, it is essential that good relationships are developed with other specialties, perhaps most importantly gastroenterology, endocrinology and dermatology. The European Society for Medical Oncology (ESMO) clinical practice guidelines, ‘Management of toxicities from immunotherapy’, contain comprehensive guidance for the use of IOs in the clinic (see Figure 2).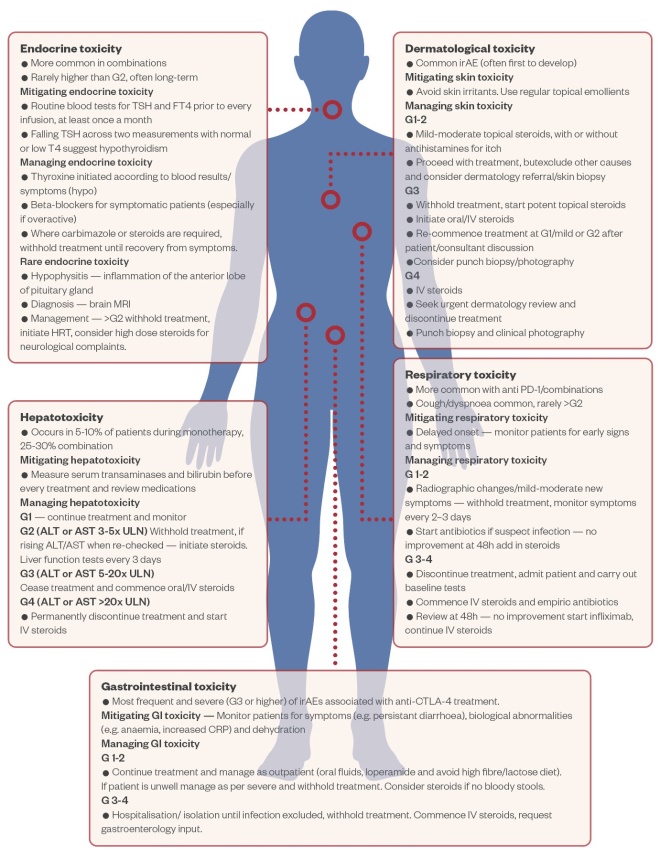
Figure 2: The most common immune-related adverse events associated with the use of immune checkpoint inhibitors
Adapted from Pharm J [86]
Abx: Antibiotics; CRP: C-reactive protein; G: grade; irAE: immune-related adverse event
Predicting the efficacy and safety of immune checkpoint inhibitors based on immune-related adverse effects
A recent study, published in 2019, has shown that patients who experience irAEs during anti-PD-1 monotherapy have a higher chance of achieving an objective response compared with those who do not experience any irAEs[97] . This provides an opportunity to predict the likely efficacy of treatment, potentially allowing more informed decisions about whether treatment should be continued in certain patients. The study involved 106 patients who were treated with either nivolumab or pembrolizumab over a two-year period; the most common irAEs were thyroid dysfunction and nephritis. The ORR for the cohort was 41.5% (n=44), but these patients represented 82.5% (n=40) of those experiencing irAEs of any grade, compared with 16.6% (n=66) who did not experience any irAEs. Furthermore, patients who experienced irAEs had significantly improved PFS than those who did not (i.e. 10 months vs. 3 months) as well as an improvement in OS (i.e. 32 months vs. 22 months), although the latter was not deemed significant on multivariate analysis[97] . In another study, clinical benefit associated with irAEs was observed in NSCLC patients receiving anti-PD-1 therapy who were shown to have autoimmune antibodies detectable prior to treatment. Investigators concluded that these autoimmune markers may help to determine the risk–benefit ratio for individual patients, allowing therapeutic benefit to be maximised while minimising irAEs[98] . Based on analysis of patient records, 48% of the 137 patients were identified as having experienced irAEs, and for those whose blood samples testing positive for autoimmune factors (i.e. rheumatoid factor, antinuclear or antithyroid antibodies), significantly higher ORRs (i.e. 41% vs. 18%) and disease control rates (i.e. 81% vs. 54%) were achieved compared with those who had tested negative. PFS was also significantly improved by median values of 6.5 months versus 3.5 months. This effect appeared to be driven primarily by patients testing positive for rheumatoid factor compared with those who tested negative, with PFS values of 10.1 months and 3.7 months, respectively[98] . A related study, published in 2019, found that patients with pre-existing antibodies were significantly more likely to experience any-grade irAEs with rates of occurrence of 60% compared with 32% in patients who tested negatively for autoimmune antibodies[99] . A more recent study in glioblastoma demonstrated a relationship between specific genetic alterations and immune expression signatures, and a tumour’s clonal evolution during treatment with anti-PD-1 therapy[100] . For example, certain non-response mutations have been identified in the PTEN gene, while some response-linked alterations have been identified in components of the MAP kinase pathway. This is a significant finding for glioblastoma therapy because PD-1 inhibitors have not provided a survival benefit for these patients to date. Initial clinical results suggest that a subgroup of patients may benefit from anti-PD-1 treatment (e.g. median survival in responders with ICPi therapy was 14 months vs. 10 months in non-responders). The results of a prospective study published in early 2020 have suggested that it may be possible to predict the risk of thyroid dysfunction (i.e. destructive thyroiditis and hypothyroidism) in patients undergoing PD-1 inhibitor therapy[101] . The study involved baseline measurements of anti-thyroid antibodies for 209 patients with re-measurement every 6 weeks for a total of 24 weeks after initiation of therapy. Thyroid dysfunction occurred in 34.1% of the 44 patients testing positive for anti-thyroid antibodies before treatment, versus 2.4% for the 165 patients testing negative at baseline. The results also support the 6–7 week presentation timeline for thyroid dysfunction as indicated in Table 6, as no new cases occurred after 24 weeks post-treatment.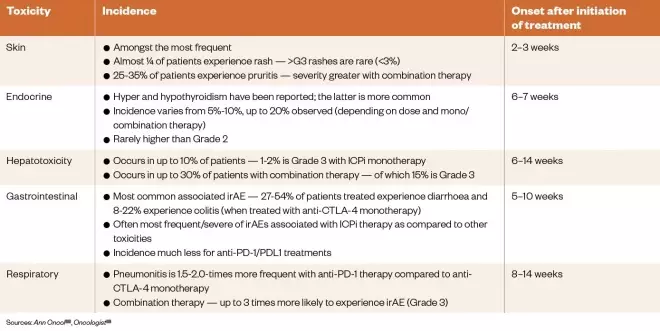
Table 6: Immune-related adverse advents associated with immune checkpoint inhibitors treatment (including incidence and onset of presentation)
Combination therapies
Patients who respond to IO monotherapy often have impressive and durable clinical responses without the side effects observed with traditional cytotoxic therapies. However, fewer than 25–50% of patients treated with ICPis fall into this population of responders. This relatively poor ORR has led to significant interest in combining ICPis with additional treatment modalities, including other IO agents, with the aim of improving response rates and durability of response. This is evident from the recent growth in combination clinical trials. For example, in the three-year period from 2014 to 2017, there was a 705% increase in the number of combination trials, but a 42% decline in enrolment sizes for individual trials, in part reflecting more targeted clinical trials. Combination therapy is attractive because it offers a means to target several mechanisms of tumour cell killing simultaneously in order to minimise tumour growth and the development of resistance[95],[102] . To date, the combination of ipilimumab with nivolumab to simultaneously target CTLA-4 and PD-1, respectively, is the only approved checkpoint inhibitor combination. It was approved in 2015 by the FDA and 2016 by the EMA for use in advanced melanoma in adults. In 2018, the FDA also approved this combination for patients with intermediate- or poor-risk, previously untreated advanced RCC[103] . While combining two ICPis is potentially associated with a higher toxicity burden, other clinical trials are now investigating whether this combination might be useful in other cancer types. Furthermore, there is a growing interest in the use of PD-1/PD-L1 inhibitors in combination with CAR-T cell therapy, given the ability of engineered T-cells to create an inflamed tumour micro-environment. The rationale for combining IO agents and chemotherapy is that the efficacies may be additive, but toxicity profiles should not overlap, potentially enhancing patient tolerability and maintaining safety. This synergy between a long-established approach to cancer treatment and a rapidly developing novel type of treatment has led to multiple approvals of chemotherapy/IO combinations, particularly for NSCLC. For example, pembrolizumab in combination with pemetrexed and platinum chemotherapy is now the first-line treatment for non-squamous NSCLC, irrespective of PD-L1 expression[104] . Another example is the combination of atezolizumab, bevacizumab, carboplatin and paclitaxel, which is now recommended as an option for metastatic non-squamous NSCLC[105] . This approach is particularly interesting, as it combines ‘conventional’ IO (i.e. targeting PD-L1) with intra-tumoural T-cell infiltration via blockade of VEGF, alongside traditional chemotherapy. However, these highly-targeted approaches can lead to more restrictive criteria for their recommendation. Interestingly, some targeted therapies such as the BRAF inhibitors are associated with some degree of immuno-modulation, and it has been postulated that their combination with IO agents may provide a synergistic effect [54],[106] . In support of this, a pre-clinical study in mice has found that the kinase inhibitor, dasatinib, significantly enhances the response to immunotherapies through its ability to inhibit the effects of the DDR2 gene, which normally helps tumours to invade healthy tissue[107] . It has been shown that depletion of DDR2 can lead to increased sensitivity of cancer cells to anti-PD-1 therapy. In the future, this may lead to a combination study with dasatinib and an anti-PD-1/anti-PD-L1 therapy in diseases such as bladder, breast and colon cancer. While radiotherapy is immunosuppressive, it promotes the release and expression of tumour neo-antigens (i.e. antigens encoded for by tumour-specific mutated genes), which results in changes to the tumour micro-environment and enhanced T-cell activity[108] . In addition, radiotherapy up-regulates PD-1 and PD-L1 expression. Both of these effects support the rationale for combining anti-PD-1/PD-L1 and radiotherapy. In particular, it is thought that ICPis should synergise with radiation-induced T-cell activity, and evaluations of this approach suggest that a clinically significant tumour response can be obtained without increased risk of toxicity compared to monotherapy[109] . Although an active area of research, the clinical use of CAR-T therapies is limited at present, and the number of clinical trials is low in comparison with other types of IO agents. For example, in 2017, there were 291 CAR-T studies reported worldwide as progressing, with 162 at the clinical stage, whereas there were 1,502 studies at the clinical stage investigating PD-1/PD-L1 agents [27] . As of mid-2018, there were 439 CAR-T combination clinical trials underway globally, of which 422 focussing on different B-cell haematological malignancies[110] ,[111] .Current challenges
The two most important challenges for IO therapies are the inability to accurately predict patient response and managing toxicities. However, the lack of information on relevant biomarkers and the high cost of research, development and treatment are also significant concerns[112] . Some observers also argue that future research should be directed towards reducing toxicity as a means to improve overall clinical benefit.Unpredictability of clinical efficacy
Newly developed agents tend to have unpredictable efficacies. There are several possible reasons for these differences in clinical responses, including the presence of different gene mutations and varying degrees of activity of specific signalling pathways in individual patients. The overall aim is to produce consistently effective agents in most patients across the majority of cancer types. Developments appear to be moving in this direction with the recent expansion of indications. For example, in 2018, the EMA expanded the marketing authorisation for pembrolizumab by adopting a new indication for the adjuvant treatment of stage III melanoma[113] . It has been suggested that the longstanding use of chemotherapy as first-line treatment for the majority of cancer types may be impeding the development and use of IO agents that are not yet widely approved for first-line use. At present, they are administered to patients who are immunocompromised owing to prior chemotherapy, so the restoration of antitumour immune function under these conditions is challenging. Therefore, it has been postulated that greater efficacies might be achieved if IO agents are utilised earlier in the treatment plan in order to utilise the full capability of the immune system[112] . Another challenge is that IO agents should ideally be directed against tumour-specific antigens solely expressed by tumour cells in order to minimise off-target effects. There would be significant clinical and economic benefits if accurate predictive biomarkers could be identified and developed, as only patients who are likely to have the greatest response would be treated. However, as seen with PD-L1 expression assays, at present there is a lack of reliability in using IO-related biomarkers to direct treatment. Another emerging challenge is the management and/or prediction of drug–drug interactions. A study reported in 2020 looking into the efficacy of atezolizumab in NSCLC patients receiving proton pump inhibitors (PPIs) and antibiotics found that patients who received PPIs or antibiotics had a poorer OS than those who did not[114] . This analysis included data from patients participating in the POPLAR and OAK trials who received either ≥2nd line atezolizumab therapy (n=757) or docetaxel therapy (n=755). Overall, 22.3% (n=169) of atezolizumab patients and 26.8% (n=202) of docetaxel patients received antibiotics, while 30.9% (n=234) and 34.4% (n=260), respectively, received PPIs 30 days before or after starting atezolizumab or docetaxel. No significant correlation between OS and the use of antibiotics/PPIs was found for the docetaxel-treated patients. However, in contrast, patients treated with atezolizumab who had also received antibiotics had an OS of 8.5 months, compared to 14.1 months for those who had not; and in PPI-treated patients, the OS was 9.6 months compared to 14.5 months for those who had not received a PPI. Overall, these results suggest that immune checkpoint efficacy can be significantly affected by some routinely prescribed drugs.Cost of immuno-oncology therapies
There are significant cost implications associated with IO-based therapies. For example, the one-year global cost of treating NSCLC with selected ICPis has been estimated at over US$80 billion[112] . The estimated cost per patient per year for a variety of IO agents is over £100,000, which places significant pressure on healthcare systems[115] . Costs for implementing these newer targeted therapies have escalated dramatically, and the duration of treatment has also lengthened because many cancer types are increasingly being treated as chronic rather than acute diseases. In the UK, the National Institute for Health and Care Excellence (NICE) is the organisation responsible for determining whether new treatments are cost-effective for the NHS. The cost of a new therapy is evaluated for its clinical effectiveness using a standardised measurement known as a quality-adjusted life year (QALY). In order to be deemed cost-effective for the NHS, a therapy should cost no more than £20,000–30,000 per QALY gained, or £50,000 for end-of-life therapies. New IO agents are increasingly exceeding these thresholds, resulting in rejection by NICE and reduced access for patients[116] . The Institute for Clinical and Economic Review, a US-based non-profit organisation providing comprehensive clinical and cost-effectiveness analyses of treatments, tests, and procedures, has studied the cost-effectiveness of the three leading immunotherapies (i.e. atezolizumab, nivolumab and pembrolizumab) and concluded that each therapy would need to be discounted by 31%–68% to reach the QALY threshold[117] . Taking this into account, NICE has stated that nivolumab cannot be recommended for routine use in the NHS with estimated QALYs of £58,791 and £78,869 versus paclitaxel and docetaxel, respectively, for treatment for urothelial cancer after cisplatin chemotherapy. NICE has also recommended that use of these agents should not be supported by the Cancer Drugs Fund (a ‘back-up’ government-sponsored fund allowing patients to obtain expensive cancer treatments through the NHS) because they do not have the potential to be cost effective[118] . Although the cost of IO agents tends to exceed QALY thresholds, consideration of the cost-effectiveness of a drug or technology is not the sole basis for decision making; clinical effectiveness and multiple patient factors are typically assessed in parallel[119] ,[120] . Often, when a new treatment strategy is evaluated, it is more clinically effective than many existing treatments, but is significantly more expensive. In this case, further economic evaluation is carried out, for example establishing the magnitude of the incremental cost-effectiveness ratio, for which an upper threshold set by NICE may not be exceeded. A decision can then be made as to whether the increase in cost is associated with an enhancement in clinical effectiveness that represents value for money [120]. Currently, there are several indications for IO agents recommended by NICE based on both cost- and clinical-effectiveness (e.g. melanoma, UC, RCC, NSCLC, lymphoma and breast cancer[121] ,[122] ,[123] ,[124],[125] ,[126],[127] ,[128],[129] ). Many pharmaceutical industry analysts have suggested that, moving forward, there should be a greater emphasis on the value and affordability of novel IO agents, rather than on generating larger numbers of potential candidates of similar therapeutic activity. There is no easy solution to this problem as it is difficult to curtail the enthusiasm of the biotechnology sector; however, it is evident that a longer-term more-sustainable research and development strategy for novel IO therapies is required. Precision medicine approaches have the potential to reduce the costs and risks associated with drug discovery and development, particularly for the clinical trials that are typically the most expensive stage of the process. The cost-saving comes from stratifying patients into smaller subsets and identifying groups that are more likely to respond, thus reducing the sizes of clinical trials and substantially reducing costs. Identifying those who are more likely to respond is also more beneficial for patients. For example, an analysis of 676 phase IIIb–IV clinical trials of NSCLC over a 14-year period found that the use of a biomarker resulted in a 26% reduction in risk-adjusted drug development costs[130] . Another option to reduce costs would be to modify treatment pathways to utilise IO agents earlier in a patient’s cancer journey, thus potentially reducing costs from treating severe ADRs often associated with conventional chemotherapy and radiotherapy, and the subsequent hospitalisation that many patients require.Future of immunotherapy
This area appears to be moving away from the development of agents selective for a given cancer type[27],[115] . IO agents are now rarely approved for one particular type of cancer; instead, there is a focus on the pathways involved and the expression of specific biomarkers in tumours, regardless of their origin or location (i.e. ‘tissue agnostic’ therapies)[131] . This pan-cancer approach is evident with the first tumour-agnostic approval of Keytruda by the FDA, in 2017, for patients with unresectable or metastatic solid tumours based on their MSI-high and dMMR status, as opposed to the location or origin of the tumour. Merck, the company which developed Keytruda, is now seeking a second pan-cancer indication against the TMB biomarker, aiming to widen patient access still further [173] . There has been a similar trend towards a tumour-agnostic approach in the small-molecule oncology area; for example, in the past two years, the kinase inhibitors larotrectinib and entrectinib have been granted accelerated approval by the FDA for use in patients with any solid tumour-type that has the NTRK fusion mutation[174] . To date, two comprehensive studies of the global IO landscape have been conducted [27] ,[115] . Over a one-year period, between September 2017 and August 2018, it was established that the global IO pipeline had increased by 67%, with cell therapy showing the most significant increase of 113% in the number of active agents, followed by other immunomodulatory (e.g. aldesleukin and interferons; 79%) and T-cell-targeted immunomodulatory therapies (76%). Importantly, the number of IO targets also increased by 50% from September 2017 to August 2018, suggesting that there could be significant broadening of the IO landscape in the future. Both reviews concluded that, of the many IO agents in clinical development, a large percentage are concentrated on only a few targets (e.g. PD-1, PD-L1 and CTLA4) [27], [115] . In addition to the five antibodies already granted FDA and EMA approval, the UK-based Cancer Research Institute has identified 164 agents in development targeting either PD-1 or PD-L1, with 50 of these at the clinical stage. This suggests that there is significant duplication in product development, and raises concerns as to whether the current approach of focusing on a small number of biomarker targets is stifling further innovation. It is noteworthy that the number of agents being developed against non-tumour-specific antigens actually decreased during the same period, consistent with the suggestion that IO is becoming too focused on a few specific targets. However, there is growing interest and enthusiasm for the IO area in both the pharmaceutical industry and academia. In addition, clinical data suggest that IO agents have significant potential for the future and may lead to several breakthrough treatments that could improve the standard of care in many different cancer types.Conclusion
IO is a fundamentally different approach to cancer therapy and is redefining the way that both solid and haematological tumours are treated. However, this new treatment paradigm is still in its infancy, and there is a long way to go in optimising the use of these novel therapies, minimising their toxicities and learning how to integrate them into the current standard of care. Furthermore, given their high cost, there are challenges ahead in incorporating them into healthcare systems in an economically sustainable manner, while increasing availability for patients. ICPis have been the focus of the recent revolution in IO, with two main antibodies (i.e. pembrolizumab and ipilimumab) receiving multiple approvals for PD-1/PD-L1 and CTLA-4 blockade, respectively. Owing to their success, there has been significant interest in combining IO agents with conventional therapies. However, despite their promising efficacy in the clinic, the ICPis produce significant toxicities in some patients. These adverse effects are frequent, but different from those seen with conventional cancer therapies. Therefore, clinical research is beginning to focus on managing and predicting these toxicities, and monitoring long-term outcomes. This should lead to guidelines on how to manage these new therapies and should encourage clinicians to use them as early as possible in treatment pathways. While the pipeline of ICPis is ever-expanding, the introduction of cancer vaccines and CAR-T cell therapies is also rapidly growing. In particular, there is a strong emphasis on developing new IO agents that can modulate T-cell activity through signalling pathways (e.g. VEGF-A, LAG-3 and IDO-1), with a view to increasing understanding of how modulation of these pathways can restore the body’s natural ability to fight cancer. The investigation of new targets and pathways in the IO area is vital to developing new therapies; however, it is important to note that combinations of presently approved IO agents with existing chemotherapeutic or biological agents are also generating significant interest. For example, a study evaluating a combination of an IO agent with an antibody-drug conjugate has reported encouraging results[132] .Disclaimer
The treatment strategies described in this review are for educational purposes only and should not be used to guide the treatment of patients. Readers are referred to National Institute for Health and Care Excellence or Scottish Intercollegiate Guidelines Network guidance in the UK, and relevant medical texts and specialist journals, for information regarding prescribing and treatment regimens.Author affiliations
Sophie Carter is a preregistration pharmacist, The Green Pharmacy, Colchester, Essex. David E. Thurston is professor of drug discovery at the School of Cancer and Pharmaceutical Sciences, Faculty of Life Sciences and Medicine, King’s College London A full PDF of this article can be found hereReferences
[1] King A & Broggio J. Cancer registration statistics, England: 2016. 2018. https://www.ons.gov.uk/releases/cancerregistrationstatisticsengland2016 (accessed May 2020)
[2] King A. Index of cancer survival for clinical commissioning groups in England: adults diagnosed 2000 to 2015 and followed up to 2016. 2017. Available at: https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/bulletins/indexofcancersurvivalforclinicalcommissioninggroupsinengland/adultsdiagnosed2000to2015andfollowedupto2016 (accessed May 2020)
[3] Cancer Research UK. Cancer survival for common cancers. 2014. Available at: https://www.cancerresearchuk.org/health-professional/cancer-statistics/survival/common-cancers-compared#heading-Three (accessed May 2020)
[4] American Cancer Society. The history of cancer. 2014. Available at: https://www.cancer.org/cancer/cancer-basics/history-of-cancer/cancer-treatment-surgery.html (accessed May 2020)
[5] Iqbal N & Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract 2014;357027. doi: 10.1155/2014/357027
[6] Decker WK da Silva RF, Sanabria MH et al. Cancer immunotherapy: historical perspective of a clinical revolution and emerging preclinical animal models. Front Immunol 2017;2(8):829. doi: 10.3389/fimmu.2017.00829
[7] Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12(4):252–264. doi: 10.1038/nrc3239
[8] American Cancer Society. Immune checkpoint inhibitors to treat cancer. 2019. Available at: https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/immunotherapy/immune-checkpoint-inhibitors.html (accessed May 2020)
[9] Mittendorf EA, Philips AV, Meric-Bernstam F et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res 2014;2(4):361–370. doi: 10.1158/2326-6066.CIR-13-0127
[10] Meyers DE, Bryan PM, Banerji S & Morris DG. Targeting the PD-1/PD-L1 axis for the treatment of non-small-cell lung cancer. Curr Oncol 2018;25(4):e324–e334. doi: 10.3747/co.25.3976
[11] Margo J. Immunotherapy: the next frontier in cancer treatment in financial review. 2017. Available at: https://www.afr.com/life-and-luxury/health-and-wellness/immunotherapy-the-next-frontier-in-cancer-treatment-20171219-h076ct (accessed May 2020)
[12] Baudino TA. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol 2015;12(1):3–20. doi: 10.2174/1570163812666150602144310
[13] O’Reilly A. ESMO e-learning: next generation drugs in kidney cancer. 2018. Available at: https://oncologypro.esmo.org/Education-Library/ESMO-E-Learning-and-V-Learning/Next-Generation-Drugs-in-Kidney-Cancer (accessed May 2020)
[14] Bristol Myers Squibb. Understanding the Science Behind Immuno-Oncology. 2019. Available at: https://www.immunooncologyhcp.com/servlet/servlet.FileDownload?file=00P1Y000017kl14UAA (accessed May 2020)
[15] Jessey T. Immunity over inability: the spontaneous regression of cancer. J Nat Sci Biol Med 2011;2(1):43–9. doi: 10.4103/0976-9668.82318
[16] Gandhi NM, Morales A & Lamm DL. Bacillus Calmette-Guerin immunotherapy for genitourinary cancer. BJU Int 2013;112(3):287–299. doi: 10.1111/j.1464-410X.2012.11754.x
[17] Cancer Research UK. What is Coley’s toxins treatment for cancer? 2012. Available at: https://www.cancerresearchuk.org/about-cancer/cancer-in-general/treatment/complementary-alternative-therapies/individual-therapies/coleys-toxins-cancer-treatment (accessed May 2020)
[18] Fuge O, Vasdev N, Allchorne P & Green JS. Immunotherapy for bladder cancer. Res Rep Urol 2015;7:65–79. doi: 10.2147/RRU.S63447
[19] Redelman-Sidi G, Glickman MS & Bochner BH. The mechanism of action of BCG therapy for bladder cancer — a current perspective. Nat Rev Urol 2014;11(3):153–162. doi: 10.1038/nrurol.2014.15
[20] Macmillan. BCG treatment for non-invasive bladder cancer. 2016. Available at: https://www.macmillan.org.uk/information-and-support/bladder-cancer/non-invasive-bladder-cancer/treating/bcg-treatment/bcg-treatment-non-invasive-bladder.html#3977 (accessed May 2020)
[21] National Institute for Health and Care Excellence. Managing non-muscle-invasive bladder cancer. 2019. Available at: https://pathways.nice.org.uk/pathways/bladder-cancer/managing-non-muscle-invasive-bladder-cancer (accessed May 2020)
[22] Cancer Research Institute. Immunotherapy treatment types. 2019. Available at: https://www.cancerresearch.org/immunotherapy/treatment-types (accessed May 2020)
[23] Geynisman DM Chien CR, Smieliauskas F et al. Economic evaluation of therapeutic cancer vaccines and immunotherapy: a systematic review. Hum Vaccin Immunother 2014;10(11):3415–3424. doi: 10.4161/hv.29407
[24] Zhang H & Chen J. Current status and future directions of cancer immunotherapy. J Cancer 2018;9(10):1773–1781. doi: 10.7150/jca.24577
[25] Galluzzi L, Vacchelli E, Bravo-San Pedro JM et al. Classification of current anticancer immunotherapies. Oncotarget 2014 Dec;5(24):12472–12508. doi: 10.18632/oncotarget.2998
[26] McKee S. EU approves first CAR-T therapies. 2018. Available at: http://www.pharmatimes.com/news/eu_approves_first_car-t_therapies_1250347 (accessed May 2020)
[27] Tang J, Shalabi A & Hubbard-Lucey VM. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol 2018;29(1):84–91. doi: 10.1093/annonc/mdx755
[28] Cristescu R, Mogg R, Ayers M et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018. 362(6411):eaar3593. doi: 10.1126/science.aar3593
[29] Teixidó C, Vilariño N, Reyes R & Reguart N. PD-L1 expression testing in non-small cell lung cancer. Ther Adv Med Oncol 2018;10:1758835918763493. doi: 10.1177/1758835918763493
[30] Sorensen SF, Zhou W, Dolled-Filhart M et al. PD-L1 expression and survival among patients with advanced non-small cell lung cancer treated with chemotherapy. Transl Oncol 2016;9(1):64–69. doi: 10.1016/j.tranon.2016.01.003
[31] Chen D, Irving B & Hodi FS. Molecular pathways: next-generation immunotherapy – inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012;18(24):6580–6587. doi: 10.1158/1078-0432.CCR-12-1362
[32] Topalian SL, Hodi FS, Brahmer JR et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690
[33] Shen X & Zhao B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: meta-analysis. BMJ 2018;362. doi: 10.1136/bmj.k3529
[34] US National Library of Medicine. Study of BMS-936558 (nivolumab) compared to docetaxel in previously treated advanced or metastatic squamous cell non-small cell lung cancer (NSCLC) (CheckMate 017). 2012. Available at: https://clinicaltrials.gov/ct2/show/NCT01642004 (accessed May 2020)
[35] US National Library of Medicine. Study of nivolumab (BMS-936558) vs. everolimus in pre-treated advanced or metastatic clear-cell renal cell carcinoma (CheckMate 025). 2012. Available at: https://clinicaltrials.gov/ct2/show/results/NCT01668784 (accessed May 2020)
[36] US National Library of Medicine. A study of atezolizumab compared with docetaxel in participants with locally advanced or metastatic non-small cell lung cancer who have failed platinum-containing therapy (OAK). 2013. Available at: https://clinicaltrials.gov/ct2/show/NCT02008227 (accessed May 2020)
[37] Dempke WCM, Fenchel K, & Dale SP. Programmed cell death ligand-1 (PD-L1) as a biomarker for non- small cell lung cancer (NSCLC) treatment – are we barking up the wrong tree? Transl Lung Cancer Res 2018;7(Suppl 3):S275–S279. doi: 10.21037/tlcr.2018.04.18
[38] Robainas M, Otano R, Bueno S & Ait-Oudhia S. Understanding the role of PD-L1/PD1 pathway blockade and autophagy in cancer therapy. Onco Targets Ther 2017;10:1803–1807. doi: 10.2147/OTT.S132508
[39] Scheel AH, Dietel M, Heukamp LC et al. Harmonized PD-L1 immunohistochemistry for pulmonary squamous-cell and adenocarcinomas. Mod Pathol 2016;29(10):1165–1172. doi: 10.1038/modpathol.2016.117
[40] Nanda S. Avelumab plus axitinib ‘new first-line standard of care’ for advanced RCC. 2018. Available at: https://oncologypro.esmo.org/Oncology-News/Daily-News/Avelumab-Plus-Axitinib-New-First-Line-Standard-Of-Care-For-Advanced-RCC (accessed May 2020)
[41] Williams, L. Pembrolizumab achieves survival improvements across PD-L1 status in KEYNOTE-407 trial. 2018. Available at: https://oncologypro.esmo.org/Oncology-News/Daily-News/Pembrolizumab-Achieves-Survival-Improvements-Across-PD-L1-Status-In-KEYNOTE-407-Trial (accessed May 2020)
[42] McLaughlin J, Han G, Schalper KA et al. Quantitative assessment of the heterogeneity of PD-L1 expression in non-small-cell lung cancer. JAMA Oncol 2016;2(1):46–54. doi: 10.1001/jamaoncol.2015.3638
[43] Arkenau HT. PD-L1 in cancer: ESMO biomarker factsheet. 2017. Available at: https://oncologypro.esmo.org/Education-Library/Factsheets-on-Biomarkers/PD-L 1-in-Cancer (accessed May 2020)
[44] Ribas A & Hu-Lieskovan S. What does PD-L1 positive or negative mean? J Exp Med 2016;213(13): 2835–2840. doi: 10.1084/jem.20161462
[45] Bassanelli M, Sioletic S, Martini M et al. Heterogeneity of PD-L1 expression and relationship with biology of NSCLC. Anticancer Res. 2018;38(7):3789–3796. doi: 10.21873/anticanres.12662
[46] Allen EMV, Miao D, Schilling B et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;9:207–211. doi: 10.1126/science.aad0095
[47] Hendriks LE, Rouleau E & Besse B. Clinical utility of tumor mutational burden in patients with non-small cell lung cancer treated with immunotherapy. Transl Lung Cancer Res 2018;7(6):647–660. doi: 10.21037/tlcr.2018.09.22
[48] Büttner R, Longshore JW, López-RÃos F et al. Implementing TMB measurement in clinical practice: considerations and requirements. ESMO Open 2019;4(1):442. doi: 10.1136/esmoopen-2018-000442
[49] Le DT et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357(6349):409–413. doi: 10.1126/science.aan6733
[50] Subrahmanyam PB, Dong Z, Gusenleitner D et al. Distinct predicitve biomarker candidates for response to anti-CTLA-4 and anti-PD-1 immunotherapy in melanoma patients. J Immunother Cancer 2018;6(1):18. doi: 10.1186/s40425-018-0328-8
[51] Tietze JK, Angelova D, Heppt MV et al. The proportion of circulating CD45RO+CD8+ memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. Eur J Cancer 2017;75:268–279. doi: 10.1016/j.ejca.2016.12.031
[52] Lutz ER, Wu AA, Bigelow E et al. Immunotherapy converts non-immunogenic pancreatic tumours into immunogenic foci of immune regulation. Cancer Immunol Res 2014;2(7):616–631. doi: 10.1158/2326-6066.CIR-14-0027
[53] Blank CU, Haanen JB, Ribas A & Schumacher TN. The “cancer immunogram”. Science 2016;658–660. doi: 10.1126/science.aaf2834
[54] Cassidy MR, Wolchok RE, Zheng J et al. Neutrophil to lymphocyte ratio is associated with outcome during ipilimumab treatment. EBioMedicine 2017;18:56–61. doi: 10.1016/j.ebiom.2017.03.029
[55] Gujar S, Pol JG & Kroemer G. Heating it up: oncolytic viruses make tumours hot and suitable for checkpoint blockade immunotherpies. Oncoimmunology 2018;7(8):e1442169. doi: 10.1080/2162402X.2018.1442169
[56] Haanen JBAG. Converting cold into hot tumors by combining immunotherapies. Cell 2017;170(6):1055–1056. doi: 10.1016/j.cell.2017.08.031
[57] Kershaw MH, Devaud C, John LB et al. Enhancing immunotherapy using chemotherapy and radiation to modify the tumor microenvironment. Oncoimmunology 201;2(9):e25962. doi: 10.4161/onci.25962
[58] Wang Y, Deng W, Li N et al. Combining immunotherapy and radiotherapy for cancer treatment: current challendes and future directions. Front Pharmacol 2018;9:185. doi: 10.3389/fphar.2018.00185
[59] Alsaab HO, Sau S, Alzhrani R et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol 2017;8:561. doi: 10.3389/fphar.2017.00561
[60] Paz-Ares L, Luft A, Vicente D et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med 2018;379(21):2040–2051. doi: 0.1056/NEJMoa1810865
[61] European Society for Medical Oncology. FDA approves pembrolizumab in combination with chemotherapy for first-line treatment of metastatic squamous NSCLC. 2018. Available at: https://www.esmo.org/oncology-news/FDA-Approves-Pembrolizumab-in-Combination-with-Chemotherapy-for-First-line-Treatment-of-Metastatic-Squamous-NSCLC (accessed May 2020)
[62] Antonia SJ, Villegas A, Daniel D et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med 2018;379(24):2342–2350. doi: 10.1056/NEJMoa1809697
[63] Antonia A, Villegas A, Vicente D et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379(24):2342–2350. doi: 10.1056/NEJMoa1809697
[64] Robert C, Long GV, Brady B et al. Nivolumab in previoulsy untreated melanoma without BRAF mutation. N Engl J Med 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082
[65] Reck M, RodrÃguez-Abreu D, Robinson AG et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774
[66] Schmid P, Adams S, Rugo HS et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 2018;379(22):2108–2121. doi: 10.1056/NEJMoa1809615
[67] Kaufman H, Sznol M, Hamid O et al. Immuno-oncology: perspectives on current therapies & future developments, in reach. 2019. Available at: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=2&cad=rja&uact=8&ved=2ahUKEwjM7NTM5tPoAhXGiVwKHXliBGcQFjABegQIBBAB&url=https%3A%2F%2Freachmd.com%2Fprograms%2Fmedical-industry-feature%2Fimmuno-oncology-perspectives-current-therapies-future-developments%2F9502%2Ftranscript%2F16996%2F&usg=AOvVaw1m6duTPEyfgWXsnNmaaNTu (accessed May 2020)
[68] Robet CM, Ribas A, Schachter J et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study.Lancet 2019;20(9):1239–1251. doi: 10.1016/S1470-2045(19)30388-2
[69] Berezina MA & Meighan-Mantha R. Immuno-oncology combination trials now easily accessible in Trialtrove. 2017. Available at: https://pharmaintelligence.informa.com/resources/product-content/immuno-oncology-combination-trials-now-easily-accessible (accessed May 2020)
[70] Buchbinder EI & Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol 2016;39(1):98–106. doi: 0.1097/COC.0000000000000239
[71] Yu JX, Hodge JP, Oliva C et al. Trends in clinical development for PD-1/PD-L1 inhibitors. Nature Reviews: Drug Discovery 2019. Available at: https://www.nature.com/articles/d41573-019-00182-w (accessed May 2020)
[72] Tang J & Yu A. PD-1/PD-L1 landscape. 2019. Available at: https://www.cancerresearch.org/scientists/clinical-accelerator/landscape-of-immuno-oncology-drug-development/pd-1-pd-l1-landscape (accessed May 2020)
[73] Solomon B & Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother 2018;67(11):1659–1667. doi: 10.1007/s00262-018-2246-5
[74] Roche. Investigational pathways: explore TIGIT. 2018. Available at: https://www.researchcancerimmunotherapy.com/investigational-pathways/explore-tigit (accessed May 2020)
[75] Stamm H, Wellbrock J & Fiedler W. Interaction of PVR/PVRL2 with TIGIT/DNAM-1 as a novel immune checkpoint axis and therapeutic target in cancer. Mamm Genome 2018;29(11–12):694–702. doi: 10.1007/s00335-018-9770-7
[76] US National Library of Medicine. A study of OMP-313M32 in subjects with locally advanced or metastatic solid tumors. 2019. Available at: https://clinicaltrials.gov/ct2/show/NCT03119428 (accessed May 2020)
[77] Bourre L. OX40 agonists: boosting cancer imcmunotherapy. 2017. Available at: https://blog.crownbio.com/ox40-ox40l-immune-co-stimulators (accessed May 2020)
[78] US National Library of Medicine. A phase I study to evaluate MEDI6383 alone and in combination with MEDI4736 in adult subjects with select advanced solid tumours. 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT02221960 (accessed May 2020)
[79] US National Library of Medicine. A phase 1b/2 safety and tolerability study of MEDI6469 in combination with therapeutic immune agents or monoclonal antibodies (MEDI6469). 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT02205333 (accessed May 2020)
[80] Corrales L, McWhirter SM, Dubensky Jr TW et al. The host STING pathway at the interface of cancer and immunity. J Clin Invest 2016;126(7):2404–2411. doi: 10.1172/JCI86892
[81] Liu M, Wang X, Wang L et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol 2018;11(1):100. doi: 10.1186/s13045-018-0644-y
[82] European Society for Medical Oncology. GITR is an attractive target for immunotherapy: targeting alternative immune pathways. 2019. Available at: https://www.esmo.org/Oncology-News/GITR-Is-an-Attractive-Target-for-Immunotherapy (accessed May 2020)
[83] Puhr HC & Ilhan-Mutlu A. New emerging targets in cancer immunotherapy: the role of LAG3. ESMO Open 2019;4:e000482. doi: 10.1136/esmoopen-2018-000482
[84] European Society for Medical Oncology. Recognising and controlling the adverse effects of CTLA-4 and PD-1 blocking agents: managing the side effects of novel cancer immunotherapies. 2014. Available at: https://www.esmo.org/Oncology-News/Managing-the-Side-Effects-of-Novel-Cancer-Immunotherapeutics (accessed May 2020)
[85] Ventola CL. Cancer immunotherapy, part 2: efficacy, safety, and other clinical considerations. P T 2017;42(7):452–463. PMID: 28674473
[86] Evans B & Evans S. Immune checkpoint inhibitors in cancer: pharmacology and toxicities. Pharm J 2018; 300(7913). doi: 10.1211/PJ.2018.20204831
[87] Wolchock JD. How is immunotherapy for melanoma changing the outlook for patients? 2018. Available at: https://www.cancerresearch.org/immunotherapy/cancer-types/melanoma (accessed May 2020)
[88] National Cancer Institute. Combination of immunotherapy drugs approved for metastatic colorectal cancer. 2018. Available at: https://www.cancer.gov/news-events/cancer-currents-blog/2018/fda-ipilimumab-nivolumab-colorectal-dna-repair (accessed May 2020)
[89] Brodsky AN. Checkpoint immunotherapy combination approved for first-line treatment of advanced kidney cancer. 2018. Available at: https://www.cancerresearch.org/blog/april-2018/fda-approval-rcc-combination-checkpoint-therapy (accessed May 2020)
[90] Haanen J, Carbonnel F, Robert C et al. Management of toxicities from immunotherapy: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2017;28(suppl 4):iv119–iv142. Available at: https://www.esmo.org/guidelines/supportive-and-palliative-care/toxicities-from-immunotherapy (accessed May 2020)
[91] Wang PF, Chen Y, Song SY et al. Immune-related adverse events associated with anti-PD-1/PD-L1 treatment for alignancies: a meta-analysis. Front Pharmacol 2017;8:730. doi: 10.3389/fphar.2017.00730
[92] Wang DY, Salem J-E; Cohen JV et al. Fatal toxic events have been characterised for patients using treatments targeting CTLA-4, PD-1 or PD-L1. JAMA Oncol 2018;4(12):1721–1728. doi: 10.1001/jamaoncol.2018.3923
[93] Larkin J, Chiarion-Sileni V, Gonzalez R et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030
[94] National Institute for Health and Care Excellence. Nivolumab with ipilimumab for untreated advanced renal cell carcinoma. Technology Appraisal guideline [TA581]. 2019. Available at: https://www.nice.org.uk/guidance/TA581/chapter/1-Recommendations (accessed May 2020)
[95] Schmidt C. The benefits of immunotherapy combinations. Nature Outlook 2017;552(7685):67–69. doi: 10.1038/d41586-017-08702-7
[96] Wang Y, Wiesnoski DH, Helmink BA et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med 2018;24(12):1804–1808. doi: 10.1038/s41591-018-0238-9
[97] Willams L. PD-1 inhibitor efficacy predicted by immune-related adverse events. 2019. Available at: https://oncologypro.esmo.org/Oncology-News/Daily-News/PD-1-Inhibitor-Efficacy-Predicted-By-Immune-Related-Adverse-Events (accessed May 2020)
[98] Nanda S. Autoimmune markers linked to immunotherapy efficacy, adverse events. 2019. Available at: https://oncologypro.esmo.org/Oncology-News/Daily-News/Autoimmune-Markers-Linked-To-Immunotherapy-Efficacy-Adverse-Events (accessed May 2020)
[99] Toi Y, Sugawara S, Sugisaka J et al. Profiling preexisting antibodies in patients treated with anti–PD-1 therapy for advanced non–small cell lung cancer. JAMA Oncol 2019;5(3):376–383. doi: 10.1001/jamaoncol.2018.5860
[100] Zhao J, Chen AX, Gartrell AD et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 2019;25(3):462–469. doi: 10.1038/s41591-019-0349-y
[101] Okada N, Iwama S, Okuji T et al. Anti-thyroid antibodies and thyroid echo pattern at baseline as risk factors for thyroid dysfunction induced by anti-programmed cell death-1 antibodies: a prospective study. Br J Cancer 2020;122(6):771–777. doi: 10.1038/s41416-020-0736-7
[102] Association of Community Cancer Centers. Immuno-oncology Institute. 2020. Available at: https://accc-iclio.org/resources/o-combination-therapy (accessed May 2020)
[103] Food and Drug Administration. FDA approves nivolumab plus ipilimumab combination for intermediate or poor-risk advanced renal cell carcinoma. 2018. Available at: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm604685.htm (accessed May 2020)
[104] National Institute for Health and Care Excellence. Lung cancer: diagnosis and management. NICE pathways. 2019. Available at: https://www.nice.org.uk/guidance/TA584/chapter/1-Recommendations (accessed May 2020)
[105] National Institute for Health and Care Excellence. Atezolizumab in combination for treating metastatic non-squamous non-small-cell lung cancer. Technology appraisal guidance [TA584]. 2019. Available at: https://www.nice.org.uk/guidance/ta584/chapter/2-Information-about-atezolizumab-plus-bevacizumab-carboplatin-and-paclitaxel (accessed May 2020)
[106] Cooper ZA, Reuben A, Amaria RN et al. Evidence of synergy with combined BRAF-targeted therapy and immune checkpoint blockade for metastatic melanoma. OncoImmunology 2014;3(9). doi: 10.4161/21624011.2014.954956
[107] Tu MM, Lee FYF, Jones RT et al. Targeting DDR2 enhances tumor response to anti-PD-1 immunotherapy. Sci Adv 2019;5(2):eaav2437. doi: 10.1126/sciadv.aav2437
[108] Mueller K. Understanding neoantigens: a critical target for improving immunotherapy. 2017. Available at: https://www.curemelanoma.org/blog/article/neoantigens-what-are-they-and-why-do-they-have-researchers-excited (accessed May 2020)
[109] Thangamathesvaran L, Shah R, Verma R et al. Immune checkpoint inhibitors and radiotherapy: concept and review of current literature. Anns of Transl Med 2018;6(8):155. doi: 10.21037/atm.2018.03.09
[110] Cell and gene therapy tracker: global CAR T-cell trials. 2018. Available at: https://www.the-scientist.com/infographics/cell-and-gene-therapy-tracker-64450 (accessed May 2020)
[111] Zhang J, Medeiros LJ & Young KH. Cancer immunotherapy in diffuse large B-cell lymphoma. Front Oncol 2018;8:351. doi: 10.3389/fonc.2018.00351
[112] Ventola CL. Cancer immunotherapy, part 3: challenges and future trends. P T 2017;42:514–521. PMID: 28781505
[113] Eurpoean Medicines Agency. Keytruda. 2018. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/keytruda (accessed May 2020)
[114] Chalabi M, Cardona A, Nagarkar DR et al. Efficacy of chemotherapy and atezolizumab in patients with non-small cell lung cancer receiving antibiotics and proton pump inhibitors: pooled post-hoc analyses of the OAK and POPLAR trials. Ann Oncol 2020;31(4):525–531. doi: 10.1016/j.annonc.2020.01.006
[115] Tang J, Pearce L, O’Donnell-Tormey J et al. Trends in the global immuno-oncology landscape. Nat Rev Drug Discov 2018;17(11):783–784. doi: 10.1038/nrd.2018.167
[116] Cancer Research UK. Health economics: the cancer drugs cost conundrum. 2018. Available at: https://www.cancerresearchuk.org/funding-for-researchers/research-features/2016-08-10-health-economics-the-cancer-drugs-cost-conundrum (accessed May 2020)
[117] Journal of Clinical Pathways. ICER report outlines TKI and PD-1 immunotherapy cost effectiveness, policy implications. 2016. Available at: https://www.journalofclinicalpathways.com/news/icer-report-outlines-tki-and-pd-1-immunotherapy-cost-effectiveness-policy-implications (accessed May 2020)
[118] National Institute for Health and Care Excellence. Nivolumab for treating locally advanced unresectable or metastatic urothelial cancer after platinum-containing chemotherapy. Technology appraisal guidance [TA530]. 2018. Available at: https://www.nice.org.uk/guidance/ta530 (accessed May 2020)
[119] Ogden J. QALYs and their role in the NICE decision-making process. 2017. Available at: https://onlinelibrary.wiley.com/doi/pdf/10.1002/psb.1562 (accessed May 2020)
[120] National Institute for Health and Care Excellence. Assessing cost effectiveness. Process and methods [PMG6]. 2019. Available at: https://www.nice.org.uk/process/pmg6/chapter/assessing-cost-effectiveness (accessed May 2020)
[121] National Institute for Health and Care Excellence. Nivolumab for adjuvant treatment of completely resected melanoma with lymph node involvement or metastatic disease. Technology appraisal guidance [TA558]. 2019. Available at: https://www.nice.org.uk/guidance/TA558 (accessed May 2020)
[122] National Institute for Health and Care Excellence. Pembrolizumab for treating locally advanced or metastatic urothelial carcinoma after platinum-containing chemotherapy. Technology appraisal guidance [TA519]. 2019. Available at: https://www.nice.org.uk/guidance/TA519/chapter/1-Recommendations (accessed May 2020)
[123] National Institute for Health and Care Excellence. Nivolumab with ipilimumab for untreated advanced renal cell carcinoma. Technology appraisal guidance [TA581]. 2019. Available at: https://www.nice.org.uk/guidance/ta581/chapter/3-Committee-discussion#cancer-drugs-fund (accessed May 2020)
[124] National Institute for Health and Care Excellence. Atezolizumab for treating locally advanced or metastatic non-small cell lung cancer after chemotherapy. Technology appraisal guidance [TA520]. 2018. Available at: https://www.nice.org.uk/guidance/ta520 (accessed May 2020)
[125] National Institute for Health and Care Excellence. Nivolumab for previously treated non-squamous non-small-cell lung cancer. Technology appraisal guidance [TA484]. 2019.Available at: https://www.nice.org.uk/guidance/ta484/chapter/4-Committee-discussion#overall-conclusions (accessed May 2020)
[126] National Institute for Health and Care Excellence. NICE approval for nivolumab provides new treatment for advanced blood cancer. 2017. Available at: https://www.nice.org.uk/news/article/nice-approval-for-nivolumab-provides-new-treatment-for-advanced-blood-cancer (accessed Apri 2020)
[127] National Institute for Health and Care Excellence. Brentuximab vedotin for treating CD30-positive cutaneous T-cell lymphoma. Technology appraisal guidance [TA577]. 2019. Available at: https://www.nice.org.uk/guidance/ta577/chapter/3-Committee-discussion#cost-effectiveness-results (accessed May 2020)
[128] National Institute for Health and Care Excellence. Advanced breast cancer – everything NICE says in an interactive flow chart: hormone receptor-positive and HER2-positive disease. NICE pathway. 2019. Available at: https://pathways.nice.org.uk/pathways/advanced-breast-cancer#path=view%3A/pathways/advanced-breast-cancer/managing-advanced-breast-cancer.xml&content=view-node%3Anodes-hrpos-and-her2pos (accessed May 2020)
[129] National Institute for Health and Care Excellence. Advanced breast cancer – everything NICE says in an interactive flowchart: triple negative disease (hormone receptor-negative and HER2-negative). NICE pathway. 2019. Available at: https://pathways.nice.org.uk/pathways/advanced-breast-cancer#path=view%3A/pathways/advanced-breast-cancer/managing-advanced-breast-cancer.xml&content=view-node%3Anodes-triple-negative-disease (accessed May 2020)
[130] Krzyszczyk P, Acevedo A, Davidoff EJ et al. The growing role of precision and personalized medicine for cancer treatment. Technology (Singap World Sci) 2018;6(3–4):79–100. doi: 10.1142/S2339547818300020
[131] Flaherty KT, Le DT & Lemery S. Tissue-agnostic drug development. Am Soc Clin Oncol Educ Book 2017;37:222–230. doi: 10.1200/EDBK_173855
[132] Bristol Myers Squibb. Seattle Genetics and Bristol-Myers Squibb highlight first data from phase 1/2 study evaluating ADCETRIS® (brentuximab vedotin) in combination with Opdivo (nivolumab) in relapsed or refractory hodgkin lymphoma at ASH annual meeting. 2016. Available at: https://news.bms.com/press-release/bmy/seattle-genetics-and-bristol-myers-squibb-highlight-first-data-phase-12-study-eval (accessed May 2020)
[133] Food and Drug Administration. Novel drug approvals for 2018. https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2018 (accessed May 2020)
[134] European Medicines Agency. 2018. Available at: https://www.ema.europa.eu (accessed May 2020)
[135] Wishart DS, Feunang YD, Guo AC et al. DrugBank 5.0: a major update to the DrugBank database for 2018. 2018. Nucleic Acids Res 2018;46(D1):D1074–D1082. doi: 10.1093/nar/gkx1037
[136] Hartley JA, Berardini M, Ponti M et al. DNA cross-linking and sequence selectivity of aziridinylbenzoquinones – a unique reaction at 5’-GC-3’ sequences with 2,5-diaziridinyl-1,4-benzoquinone upon reduction. Biochemistry 1991;30(50):11719–11724. doi: 10.1021/bi00114a016
[137] Demko S, Summers J, Keegan P et al. FDA drug approval summary: alemtuzumab as single-agent treatment for B-cell chronic lymphocytic leukemia. Oncologist 2008;13:167–174. doi: 10.1634/theoncologist.2007-0218
[138] Jackson C, Hartley JA, Jenkins TC et al. N-2,N-4,N-6-tri(hydroxymethyl)-N-2,N-4,N-6-trimethylmelamine (trimelamol) is an efficient DNA cross-linking agent in vitro. Biochem Pharmacol 1991;42(11):2091–2097. doi: 10.1016/0006-2952(91)90343-4
[139] Jackson C, Crabb TA, Gibson M et al. Studies on the stability of trimelamol, a carbinolamine-containing antitumor drug. J Pharm Sci 1991;80(3):245–251. doi: 10.1002/jps.2600800311
[140] Morris SJ, Thurston DE & Nevell TG. Evaluation of the electrophilicity of DNA-binding pyrrolo[2,1-c][1,4]benzodiazepines by HPLC. J Antibiotics (Tokyo) 1990;43(10):1286–1292. doi: 10.7164/antibiotics.43.1286
[141] Bose DS & Thurston DE. Boron trifluoride promoted cleavage of benzyl carbamates. Tetrahedron Letters 1990;31(47):6903–6906. doi: 10.1016/S0040-4039(00)97203-4
[142] Bristol Myers Squibb. Yervoy – US full rescribing information. 2018. Available at: https://packageinserts.bms.com/pi/pi_yervoy.pdf (accessed May 2020)
[143] Cancer Research Institute. Timeline of progress. 2018. Available at: https://www.cancerresearch.org/immunotherapy/timeline-of-progress (accessed May 2020)
[144] Imperial Cancer Research Fund General Practice Research Group. Effectiveness of a nicotine patch in helping people stop smoking: results of a randomised trial in general practice. BMJ 1993;306(6888):1304–1308. doi: 10.1136/bmj.306.6888.1304
[145] European Society for Medical Oncology. OncologyPRO news 2018. 2018. Available at: https://oncologypro.esmo.org (accessed May 2020)
[146] Postow MA & Wolchok J. Ipilimumab: developmental history, clinical considerations and future perspectives. The Melanoma Letter 2012;30:1–4. Available at: https://www.medscape.com/viewarticle/762282_5 (accessed May 2020)
[147] FDA expands approval of Ipilimumab to include paediatric patients 12 years and older with unresectable or metastatic melanoma. 2017. Available at: https://www.esmo.org/Oncology-News/FDA-Expands-Approval-of-Ipilimumab-to-Include-Paediatric-Patients-12-Years-and-Older-with-Unresectable-or-Metastatic-Melanoma (accessed May 2020)
[148] Alexander W. The checkpoint immunotherapy revolution. P T 2016;41(3):185–191. PMID: 26957887
[149] Dharma R. AstraZeneca’s tremelimumab receives FDA approval to treat mesothelioma. 2015. Available at: https://www.pharmaceutical-technology.com/news/newsastrazenecas-tremelimumab-receives-fda-approval-treat-mesothelioma-4556286 (accessed May 2020)
[150] Bristol Myers Squibb. Our company – history timeline FDA approves OPDIVO. 2014. Available at: https://packageinserts.bms.com/pi/pi_opdivo.pdf (accessed May 2020)
[151] EMA recommends pembrolizumab for the adjuvant treatment of melanoma. 2018. Available at:https://www.esmo.org/Oncology-News/EMA-Recommends-Pembrolizumab-for-the-Adjuvant-Treatment-of-Melanoma?hit=mail-snews&utm_campaign=Scientific&utm_source=hs_email&utm_medium=email&utm_content=67114737&_hsenc=p2ANqtz-_XbL6ziYc79SkP7-VI-HOTUdVKYA9cKH4VjpOfuiOPcpOZbVOI2q_lkNTfCC2rBBBZLjPg1b4h8p3tcTy2sAUzFomTUw&_hsmi=67114737 (accessed May 2020)
[152] Hu Y, Turner MJ, Shields J et al. Investigation of the mechanism of action of alemtuzumab in human CD52 transgenic mouse model. Immunology 2009;128(2):260–270. doi: 10.1111/j.1365-2567.2009.03115.x
[153] Plieth J. Antibody-drug conjugate resurgence continues with Lumoxiti. 2018. Available at: https://www.evaluate.com/vantage/articles/news/snippets/antibody-drug-conjugate-resurgence-continues-lumoxiti (accessed May 2020)
[154] Chalouni C & Doll S. Fate of antibody-drug conjugates in cancer cells. J Exp Clin Cancer Res 2018;6;37(1):20. doi: 10.1186/s13046-017-0667-1
[155] Kreitman RJ & Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res 2011;17(20):6398–6405. doi: 0.1158/1078-0432.CCR-11-0487
[156] European Medicines Agency. Adcetris: summary for the public. 2012. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/adcetris (accessed May 2020)
[157] Food and Drug Administration. Adcetris drug approval package. 2011. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125399_adcetris_toc.cfm (accessed May 2020)
[158] Food and Drug Administration. Approval date(s) and history, letters, labels, reviews for BLA 125427. 2013. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=125427 (accessed May 2020)
[159] European Medicines Agency. Kadcyla: summary for the public. 2013. Available at: https://www.ema.europa.eu/documents/overview/kadcyla-epar-summary-public_en.pdf (accessed May 2020)
[160] European Medicines Agency. Besponsa: summary for the public. 2017. Available at: https://www.ema.europa.eu/documents/overview/besponsa-epar-summary-public_en.pdf (accessed May 2020)
[161] Food and Drug Administration. Approval date(s) and history, letters, labels, reviews for BLA 761040. 2017. Available at: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event =overview.process&ApplNo=761040 (accessed May 2020)
[162] European Medicines Agency. Mylotarg: summary for the public. 2018. Available at: https://www.ema.europa.eu/documents/overview/mylotarg-epar-summary-public_en.pdf (accessed May 2020)
[163] Food and Drug Administration. FDA approves gemtuzumab ozogamicin for CD33-positive AML. 2018. Available at: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm574518.htm (accessed May 2020)
[164] Alva A, Daneils GA, Wong MKK et al. Contemporary experience with high-dose interleukin-2 therapy and impact on survival in patients with metastatic melanoma and metastatic renal cell carcinoma. Cancer Immunol Immunother 2016;65(12):1533–1544. doi: 10.1007/s00262-016-1910-x
[165] Wadler S & Schwartz EL. New advances in interferon therapy of cancer. Oncologist 1997;2:254–267. PMID: 10388057
[166] Zhao W, Schorey JS, Bong-Mastek M et al. Role of a bacillus Calmette-Guérin fibronectin attachment protein in BCG-induced antitumor activity. I Int J Cancer. 2000;86(1):83–88. doi: 10.1002/(SICI)1097-0215(20000401)86:1<83::AID-IJC13>3.0.CO;2-R
[167] Anassi E & Ndefo UA. Sipuleucel-T (provenge) injection: the first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. P T. 2011;36(4):197–202. PMID: 2157277
[168] Fukuhara H, Ino Y & Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci 2016;107(10):1373–1379. doi: 10.1111/cas.13027
[169] Filley AC & Dey M. Immune system, friend or foe of oncolytic virotherapy? Front Oncol 2017;7:106. doi: 10.3389/fonc.2017.00106
[170] Kohlhapp FJ & Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clini Cancer Res 2016;22(5):1048–1054. doi: 10.1158/1078-0432.CCR-15-2667
[171] González-RodrÃguez E & RodrÃguez-Abreu D. Immune checkpoint inhibitors: review and management of endocrine adverse events. Oncologist 2016;21(7):804–816. doi: 10.1634/theoncologist.2015-0509
[172] Pishvaian MJ, Blais EM, Brody JR et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol 2020;21(4):508–518. doi: 10.1016/S1470-2045(20)30074-7
[173] Ray T. Merck bid for second pan-cancer Keytruda indication would raise tumor mutation burden to CDx status. 2020. Available from: https://www.genomeweb.com/molecular-diagnostics/merck-bid-second-pan-cancer-keytruda-indication-would-raise-tumor-mutation (accessed May 2020)
[174] Gray A. Going against type: the new class of cancer therapies targeting mutations rather than tissues. Pharm J 2020;304(7935):164–166. doi: 10.1211/PJ.2020.20207799
[175] Kotch C, Barrett D & Teachey DT. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev Clin Immunol 2019;15(8):813–822. doi: 10.1080/1744666X.2019.1629904