

- Introduction of the ‘precision medicine’ approach has seen its most significant advances in the field of oncology.
- The key scientific principle behind this approach is to identify predictive biomarkers that can be used to select the most suitable therapeutic agents.
- Biomarkers can also be used for diagnostic, prognostic, toxicological and treatment monitoring purposes.
- Many pharmacogenomic assays and kits have been developed to identify biomarkers.
Cancer learning ‘hub’
Pharmacists are playing an increasingly important role in supporting patients with cancer, working within multidisciplinary teams and improving outcomes. However, in a rapidly evolving field with numbers of new cancer medicines is increasing and the potential for adverse effects, it is now more important than ever for pharmacists to have a solid understanding of the principles of cancer biology, its diagnosis and approaches to treatment and prevention. This new collection of cancer content, brought to you in partnership with BeOne Medicines, provides access to educational resources that support professional development for improved patientIntroduction
Each individual has their own unique genome, which can include small single nucleotide polymorphisms (SNPs) and/or large changes in DNA base pair sequence (mutations). These can be inherited but can also be introduced during a person’s lifetime through external agents (e.g. carcinogenic chemicals or radiation). Although usually harmless to the well-being of the individual, these genetic modifications can affect the way the body responds to a therapeutic agent either through differences to the drug target, or through ADMET considerations (i.e. absorption, distribution, metabolism, excretion and toxicology). Precision medicine, sometimes known as ‘personalised medicine’[1],[2] and abbreviated to ‘PM’, is a term that is increasingly being used to describe treatments, including therapeutic agents, tailored to individual patients or groups of patients[3] . The overall goal is to match therapies to individuals to ensure that they receive effective treatment with minimal toxicity. This is particularly important for cancer patients who may have a limited life expectancy. Furthermore, there has been an ongoing decrease in the cost of sequencing the human genome, which has led to the widespread adoption of integrative sequencing strategies for the study of cancer and the PM approach[4] . Although often used interchangeably with personalised medicine, the term ‘precision medicine’ was first introduced by the US National Research Council in 2011 with the aim of conveying a broader concept that, although it may not be possible to develop treatments specifically for individual patients on a one-off basis, it should be feasible to at least define subgroups of patients and target them through a genomics-based approach[5] . The concept of PM in oncology gained further momentum when the PM Initiative was launched in the US in 2015, accelerating the development of biomarker-driven therapeutic strategies[6] . Within the field of oncology, the most significant aspect of a PM approach involves the identification of a ‘biomarker’ associated with a particular cancer type. A biomarker is a unique mutated nucleic acid sequence, protein, glycoprotein or group of proteins, expressed by the tumour cells but not normally by healthy cells[1] . There are four main types of biomarkers: pre-disposition (indicating the likelihood of developing the disease), diagnostic (used to confirm the patient has a particular cancer), predictive (determining which cohort of patients may benefit from a particular drug therapy) and prognostic (suggesting how the cancer may develop in the individual)[7] . Each biomarker type is relevant at different stages of the disease (see Figure 1)[1] .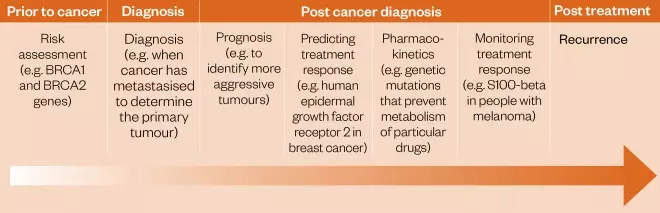
Figure 1: Role of biomarkers in oncology
Technologies used for diagnostic testing
Companion diagnostic (CDx) tests are based on a number of different platform technologies, each with their own advantages and limitations:- Immunohistochemistry (IHC): IHC is based on visualisation of the interaction between antibodies and antigens, a process that can discriminate between abnormal (e.g. cancerous) and healthy cells and tissues[13] . Although IHC can effectively visualise the localisation of different antigens and biomarkers [13] , it has a number of limitations, the main one being intra-observer variability in identifying the intensity and extent of the true cellular staining[13] .
- In situ hybridisation (ISH): this uses fluorescent nucleic acid probes to detect, bind to, identify, interpret and map mutations in genes, or regions of genes, that frequently appear only in cancer cells[14],[15] . Most tests of this type have the limitation that only single defined mutations are detected, whereas cancer cells can develop multiple mutations[16] .
- Immunoassays: based on antibody/antigen recognition, these assays are capable of detecting a range of biomarkers including those related to toxicity, the levels of active drug metabolites in the blood, and the overall levels of drugs for therapeutic monitoring purposes. In general, serum marker testing kits may be non-specific for different cancer types[17] .
- Polymerase chain reaction (PCR): based on DNA amplification, this technology can detect single-base changes within genes, making it a favourable option for the detection of gene-based cancer biomarkers[18] . However, degradation of the sample being analysed may increase the number of false-negative results[19] .
- Flow cytometry: this uses a laser- or impedance-based biophysical technology for cell sorting and counting, and biomarker detection is achieved by passing a suspension of cells through an electronic detection device. This allows simultaneous multiparametric analysis of the chemical and physical characteristics of thousands of cells per second[20] . Flow cytometry preserves the cells for further studies but requires multiple fluorophores to differentiate cell types, and this can lead to large amounts of data that can be difficult to analyse and interpret[20] .
- Tandem mass spectrometry: this methodology, especially if interfaced with liquid, gas or capillary chromatography systems, can be used to accurately measure the levels of both biomarkers and drugs in tissue samples[21] .
- Next-generation sequencing (NGS): this is a high-throughput method for screening short or long DNA sequences, as well as identifying biomarkers that are highly expressed. Although the technology is relatively expensive, its costs are reducing, and it is an increasingly recognised method for diagnosing, predicting risk and classifying different cancer types[22] . For example, the FDA has issued two guidance documents to drive the use of NGS as a diagnostic method for identifying the risk of developing a genetic disease[23] . The first provides CDx test developers with FDA-approved public databases containing clinical evidence that can corroborate the accuracy of NGS-based genomic testing results[23] ,[24] . The second document offers recommendations to potential manufacturers on how to develop NGS testing, highlighting key elements that the FDA will look for in a pre-market submission when assessing an assay’s analytical validity[23] ,[25] .
Limitations of biomarker testing
The NCI Dictionary of Cancer Terms defines a biomarker as a biological molecule found in blood, other body fluids or tissues that is a sign of a normal or abnormal process, or of a condition or disease; or that may be used to see how well the body responds to a treatment for a disease or condition[26] . In this review, the focus is on biomarkers that can be used to select which patients are most likely to respond to particular anticancer therapies. Although simple in concept, in practice there are a number of difficulties and limitations, some of which have been previously reviewed[27] . The most significant problems and limitations are briefly described below.Acceptability
Whereas blood samples are relatively easy to obtain from patients with haematological cancers, not all solid tumours are straightforward to biopsy. For example, biopsies can more easily be carried out on tumours near to the skin surface (e.g. skin, bladder, breast, prostate, oesophaegal and bowel cancers), but for organs situated deeper in the body (e.g. liver, pancreas, lungs) biopsies normally require major surgery. In terms of risk and tolerability for the patient, blood or urine samples are lowest risk and well tolerated. Solid tumour and cerebrospinal fluid biopsies are higher risk in terms of infections and secondary tissue damage, and are less well-tolerated by patients.Measurement errors
Within the context of this review, the search for a biomarker is usually made in tumour cells obtained from biopsy material taken from solid tumours, or from blood samples in the case of haematological malignancies. There are a number of potential measurent errors associated with sampling, extraction and analysis. For example, with solid tumours that are comprised of multiple clones of cells at different stages of cancer evolution, it is possible that the small sample of cells removed at biopsy do not contain the relevant biomarkers that may be present in cells at other sites within the tumour mass. This is less of a problem with haematological cancers, where a relatively large blood sample is likely to contain at least some of the biomarker-relevant cancer cells. Other errors can result from imperfect extraction and analytical techniques, especially when working with very small samples from solid tumour biopsies.Sampling bias
This can occur at different stages of the biomarker measuring process, from selecting patients to storing and measuring samples. For example, there may be a bias in sampling patients whose tumours are more convenient for sampling, and also in sampling parts of solid tumours closer to the surface of the body.Confounding errors
These can arise through failure to identify either internal (e.g. a patient’s weight) or external (e.g. suitable laboratory equipment) factors that can affect the amount of evaluable biomarker reported in a sample.Role of regulatory bodies validating biomarkers and CDx tests
The development of a CDx assay kit in conjunction with a new drug depends on when the biomarker is first identified[28] . If a biomarker is first discovered during the early stages of the patient selection process, then the process of assay development can begin early[28] . However, if the biomarker is discovered as a response to therapy, development of the diagnostic assay will generally occur at a later stage[28] . In 2016, the FDA released draft guidance on the ‘Principles for codevelopment of an in vitro companion diagnostic device with a therapeutic product’[29] , which aimed to be a practical guide for companies that are developing treatments that rely on a biomarker test for their use. The FDA has recognised that simultaneous development and launch of a drug and corresponding biomarker assay may not always be feasible, therefore this document provides advice for pharmaceutical companies on this issue[29] . Although there are some biomarkers that have been specifically approved by the FDA for use in disease monitoring (e.g. nuclear matrix protein-22 for bladder cancer), these biomarkers are not usually specific enough to be used for general population screening[28] . CDx tests are classed as medical devices that provide essential information for the safe and effective use of medicines[30] . According to the FDA, their three main roles are: identifying patients who are likely to benefit from a particular therapeutic product, to help identify those who are likely to be at an increased risk of developing side effects from a particular medication, and to monitor and adjust a patient’s response to treatment. In oncology, CDx tests are mainly used for identifying patients who are expressing a particular biomarker to select a suitable treatment, and to identify patients at high risk of developing a side effect from a given drug therapy[26] . The FDA approves CDx tests through the Center for Devices and Radiological Health (CDRH), which verifies both the analytical and clinical validation of in vivo testing kits[31] . Within the EU, regulation of medicinal devices has been separated from the regulation of pharmaceuticals. The EMA has taken a less active role in regulating companion diagnostics, as the kits are regarded as in vitro diagnostics (IVDs)[32] . Therefore, the EMA does not have complete authority over their regulation[32] . According to European directive 98/79/EC[32],[33] , CDx tests that are considered to be an IVD medicinal device and have the ‘CE’ mark can be distributed to all members of the European Economic Area[32] . However, the EU is planning a substantial reform of their current regulation rules to strengthen the system to incorporate new CDx tests into the regulatory framework[32] . Recently, the EMA launched a consultation paper on the development and lifecycle of PMs and their CDx tests[34] .Precision medicine-based clinical trials
To evaluate the PM approach more thoroughly, and to attempt to match targeted therapies to genomic profiling, several clinical trials have been launched in the US. These have been reviewed by Gong et al. [35],[32] and include the Signature, I-SPY, National Cancer Institute (NCI)-sponsored NCI-MATCH, NCI-MPACT, ALCHEMIST, Lung-MAP, Pediatric MATCH, Exceptional Responders, Lung Cancer Mutation Consortium (LCMC)-sponsored, and the American Society of Clinical Oncology (ASCO)-sponsored TAPUR (Targeted Agent and Profiling Utilization Registry) clinical trials[31] . These non-randomised clinical trials are considered to be important for the future of PM approaches, and are aimed at evaluating the performance (both efficacy and safety) of FDA-approved targeted anticancer agents prescribed for the treatment of patients with advanced cancer and with a potentially actionable genomic alteration[36] . According to ASCO, more than 1,700 participants have consented to participate, and more than 1,220 have been treated with a TAPUR study drug. Clinicians who order the 596-gene Tempus xT assay (Tempus Labs, Inc) receive a report that has been optimised for TAPUR participation with a specific summary of the genomic alterations targeted by study drugs, which helps clinicians screen for trial-eligible patients. Importantly, a number of pharmaceutical companies collaborated to contribute to these trials, and the choices of genomic profiling tests requested by participating clinical oncologists are catalogued with the aim of identifying signals of drug activity. For example, a molecular aberration such as BRAF V600E seen in any cancer type can be matched for treatment with BRAF inhibitors (which are presently only FDA-approved for melanoma and a few other specific histologies). In another example, clinical trials are identifying tumours of any type harbouring mutations in any of the Fanconi anaemia pathway genes, most commonly found in tumours with a mutation pathway in BRCA1/2 [37],[38] , to match patients with PARP inhibitors. In the UK, Innovate UK recently launched a €6m initiative for the development of PM technologies that can be used to lead and improve targeted therapies.Sources and selection criteria
Cancer-relevant biomarkers, the assays to detect and evaluate them, and the associated anticancer agents are reviewed below based on information from the primary and patent literature, and from material available from conferences and the websites of pharmaceutical companies, medical charities and organisations such as the Medicines and Healthcare products Regulatory Agency (MHRA), the European Medicines Agency (EMA), the FDA, the UK government’s National Institute for Health and Care Excellence (NICE)[39],[40] the British National Formulary (BNF), the Journal of Precision Medicine, and web-based resources such as GenomeWeb. Although the information has been grouped in sections relating to cancer type, some biomarkers are highly specific for a particular type (e.g. BCR-ABL for chronic myeloid leukemia), whereas others are associated with more than one tumour type (e.g. BRCA1/2 in breast, ovarian and prostate cancers)[41] .Biomarkers, CDx tests and PM anticancer therapies in current use
This part of the two-part review will focus on solid tumours, which are defined as abnormal masses of tissue that do not contain any cysts or liquid areas[42] , and are classified histologically into sarcomas, carcinomas or lymphomas[43] . They can be further classified molecularly through the use of processes such as genome sequencing, gene-expression profiling and miRNA analysis, along with other parameters to assess them, for example DNA copy number, chromosomal instability and gene functionality[44] .Breast cancer
This is a heterogeneous disease with varied morphological and molecular characteristics[45] . One of the best ways of distinguishing between the different types of breast cancer is still through histological assessment[45] . Breast cancers are initially characterised as invasive or non-invasive and, can be further subdivided into carcinomas of no special type (ductal carcinoma) or of a special type, which includes lobular growth patterns and altered differentiation[45] . Lobular breast cancer in situ, in particular, can be detected through mammography[45] . Women are offered surgery, radiotherapy or drug treatments (or a combination of one or more of these) as part of their care plan. Biomarkers are increasingly important in the diagnosis, prognosis and treatment of breast cancer, and the most significant ones are summarised in Table 1 along with the CDx tests available and the associated anticancer agents that can be selected on the basis of the presence of a particular biomarker. Progress in this area continues, and recent research has suggested that it may be possible to identify SNPs specific for either lobular or oestrogen receptor (ER)-positive ductal breast cancers[46] . The first biomarker to be linked to breast cancer was the estrogen receptor (ER), which was first identified in the 1960s and has been used in the clinical management of this disease since the mid-1970s for diagnostic, prognostic and predictive treatment purposes. ER-positive tumours comprise up to 75% of all breast cancers diagnosed, representing 65% and 80% of patients aged under and over 50 years respectively[47] . It is also most commonly found in patients with invasive ductal cancers. Both ER and progesterone receptor (PgR) status are routinely assessed for all invasive breast cancers using IHC methods, and the results reported in a standardised way known as the Allred system. This method scores cells based on an estimated proportion and intensity of staining on a scale of 0 to 8, where a score of 3 or greater is defined as positive[48] . Patients with ER/PgR-positive breast cancer are treated with endocrine therapies such as selective estrogen-receptor modulators (SERMs) (e.g. tamoxifen) or aromatase inhibitors (e.g. anastrazole) in both the adjuvant and metastatic settings. PgR is used as a predictive biomarker for ER-α function and breast cancer prognosis[49] . In the presence of an agonist ligand, PgR becomes associated with ER-α, directing chromatin-binding events within breast tumour cells, and both receptor and hormone levels can influence the interaction between ER-α and PgR[49] . This cross-talk is involved in regulating the gene expression programme associated with low tumourigenicity, and so is associated with a more favourable outcome for the patient[49] . Although this is a potentially useful biomarker in breast cancer, NICE guidelines state that it should not be used routinely to assess the prognosis of patients with invasive breast cancers[50] . IHC assays are used to assess the level of PgR in tumours[51] . Gene amplification, or protein over-expression of human epidermal growth factor receptor 2 (HER2), discovered in the 1980s[52] , is often associated with poor prognosis in breast cancer. However, it can be used to identify patients who may benefit from targeted therapies such as the antibody trastuzumab (Herceptin , Roche) or the antibody–drug conjugate (ADC) trastuzumab emtansine (Kadcyla , Roche). HER2 is over-expressed in around 15% of cases of early invasive breast cancer, and around 77% of those that are also ER-positive[53],[54] . Diagnostic tests for HER2 over-expression and gene amplification include IHC and ISH methods, and standardised quality-assured methodologies have been developed[55],[56] . The results are reported as HER2 negative or HER2 positive according to NICE guidelines[55],[56] . The results are reported as HER2 negative or HER2 positive according to NICE guidelines, with test results scoring 3+ by IHC, or 2+ and amplified by ISH, counted as positive. Mutated versions of the breast cancer genes 1 and 2 (BRCA1 and BRCA2), although found in only a small percentage of the general population[57] , are classified as high-risk mutated genes, and are the best-known predicative genetic biomarkers for both breast and ovarian cancer[58] . BRCA1 and BRCA2 are tumour suppressor genes that have been shown to be pivotal in many cellular processes, including the transcriptional regulation of DNA after damage, as well as DNA repair[59] . In addition, they are involved in protecting the genome from damage through the BRCA proteins that maintain chromosomal stability[59] . Mutations in these genes can be passed through generations in both sexes[58] . However, a recent study has suggested that young breast cancer patients with BRCA mutations have a similar survival rate to those without the mutation[60] . This is an important finding as, prior to this study, early diagnosis of this mutation frequently led to elective double mastectomies in healthy patients as a prophylactic measure, and in risk-reducing bilateral salphingo-oophrectomy in ovarian cancer patients after chemotherapy treatment[60] . The CDK4 and CDK6 kinases associated with Cyclin D1 are over-expressed in all subtypes of breast cancers[61] . The presence of both CDK4 and Cyclin D1 are needed to maintain tumour cell proliferation[61] . While the Cyclin D1/CDK4 complex has two functions, the main one is to sustain the tumourigenic potential of breast cancer cells, although researchers agree that more studies are required to fully explain why the Cyclin D1/CDK4 complex plays a critical role in tumour cells but is less important in normal cells[61] . Drugs such as palbociclib (Ibrance , Pfizer), ribociclib (Kisqali; Novartis) and abemaciclib (Verzenio; Eli Lilly) are small-molecule inhibitors of CDK4 and CDK6, with a high selectivity for these kinases compared with other cyclin-dependent kinases. Although no CDx tests are available, for these agents, they appear to work more effectively in patients with hormone-receptor-positive breast cancer, and are synergistic with other endocrine therapies[62] . PIK3CA mutations with associated over-activation of the PI3K signalling pathway were identified by Novartis scientists as associated with tumour growth, resistance to endocrine-based therapies and relatively poor treatment outcomes[63],[64] . A recent study (i.e. SOLAR 1) analysed breast cancer tissue and circulating tumour DNA in 572 patients, identifying PIK3CA mutations in 40% of hormone receptor positive or negative breast cancers[63],[64] . The kinase inhibitor alpelisib (Piqray) was developed by Novartis to target PIK3CA mutations, and is approved by the FDA for use in breast cancer[63],[64] . It is used alongside Qiagen’s Therascreen PIK3CA RGQ PCR test to select patients suitable for treatment[63],[64] . Development of a similar kinase inhibitor (i.e. taselisib) by Roche to target PIK3 mutations for the treatment of metastatic breast cancer was halted due to a lack of efficacy and unacceptable toxicity[65] . In addition to OncotypeDX , other test kits that profile panels of genes to assist with clinical decision making in breast cancer include MammaPrint (Agendia), EndoPredict (Myriad Genetics Inc), IHC4, Mammostrat (Clarient Diagnostic Services) and Prosigna (NanoString Technologies Inc). However, none of these have been recommended for use in the UK by NICE[66] , which has produced several draft reports weighing evidence on the use of diagnostic testing kits of this type[67] .Lung cancer (small cell and non-small cell)
EGFR plays a critical role in regulating normal cell proliferation, apoptosis and other cellular functions. Around 10% of non-small cell lung cancer (NSCLC) patients in the UK and 35% in East Asia have EGFR mutations associated with their tumours[74] . Initial clinical studies carried out by Riley et al. showed that first-generation tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib gave a significant clinical response in 10% of Caucasian and 25–30% of Asian patients[75] . However, patients who responded well initially soon developed resistance due to secondary acquired EGFR mutations (75), the main one of which was a change of threonine to methionine at amino acid position 790 (i.e. T790M) in exon 20 of the EGFR gene[76] , resulting in a reduced binding affinity for these first-generation TKIs[76] . This understanding of the mechanism of action allowed researchers to develop improved TKI molecules, such as osimertinib (a third-generation TKI)[76],[77] . A number of companion diagnostic tests have been developed to identify patients with EGFR biomarkers, and some of these have been approved for use in the NHS (see Table 2). EML4-ALK (echinoderm microtubule-associated protein-like 4 — anaplastic lymphoma kinase) is a mutated translocation fusion gene resulting from an inversion within the short arm of chromosome 2 where ALK joins with EML4[78] . Patients who are ALK+ can be treated with TKI agents specifically designed to target this mutation. The first-generation of these was crizotinib (Xalkori; Pfizer), although further generations of ALK-1 inhibitors have since been developed. ROS-1 is a receptor tyrosine kinase encoded by the ROS-1 gene. It has structural similarity to the ALK protein but the roles of the ROS-1 protein in normal development, and the identity of its physiologic ligand, are presently uncertain. It is classed as an oncogene because it can rearrange into the ROS-1 fusion mutation, which is commonly seen in young non-smoking female patients of Asian ethnicity with adenocarcinoma histology. Crizotinib has been approved by the FDA and NICE as a viable therapy for patients whose tumours have a ROS-1 translocation[78] . CDx tests for both ALK and ROS-1 biomarkers are approved for use in the NHS (see Table 2). Vascular endothelial growth factor (VEGF 1–3), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) are all secreted by both normal and tumour cells, and induce angiogenesis by specifically binding to their respective receptors[79] . Angiogenic pathways are very important for the development of NSCLC as they promote tumour progress by facilitating the growth of new blood vessels[74] . Therefore, these three growth factors, which are involved in the regulation of angiogenesis, can be used as biomarkers for drug discovery and patient selection. The VEGF inhibitor bevacizumab (Avastin; Roche) was one of the first agents to be developed in this area, and is licenced for use in NSCLC in combination with a platinum-based therapy for the first-line treatment of NSCLC tumours harbouring VEGF as a biomarker. However, it is not recommended by NICE for this purpose based on a lack of evidence (NICE Technical Appraisal TA148)[74] . PDGF plays an important role in the development of normal cells, and is essential for cell proliferation, migration, differentiation and survival. There are two main receptor isoforms (i.e. α and β), and both are associated with poor prognosis in lung cancer. It is also involved in angiogenesis, and regulates VEGF expression in tumour cells, which can progress the development of pre-cancerous NSCLC into a more advanced stage. One of the most effective ways of blocking PDGF activity is through the inhibition of intracellular PDGFR kinases. The FGF family of growth factors is involved in numerous cellular processes, one of which is angiogenesis[80] . FGF-2 is one of the most potent angiogenic factors within the family, and high expression of this ligand is associated with transformation of normal cells into malignant ones[80] . The specific role of FGF-2 in NSCLC development and progression is largely unknown[80] , although one study has established a potential role of FGFR-dependent signaling as a component of the resistance mechanism for EGFR tyrosine kinase inhibitors[81] . Both FGFR and VGFR are structurally similar, and so novel inhibitors of both are at the research stage as targeted anti-angiogenesis agents[81] . FGFR inhibitors could potentially be used either alone or in combination with other TKIs as a means of expanding the portfolio of therapies that can be used for NSCLC patients[81] . The BRAF V600E and BRAF V600K biomarkers (important in melanoma — see ‘Metastatic malignant melanoma’) are present in 1–2% of lung adenocarcinomas in patients who do not have common lung tumour mutations such as EGFR, EML4-ALK and ROS-1 [82] . The Oncomine Dx Target Test (Thermo Fisher Scientific) based on next-generation sequencing was approved to test for BRAF, ROS-1 and EGFR mutations in these patients[82],[83] . NGS panels have also been used in patients with NSCLC to detect a range of genomic alterations, including the BRAF V600E/K mutations[84] . There has been some success in measuring PD-L1 over-expression in lung tumours to predict the possible outcome of treatment with the immunotherapy agent pembrolizumab (Keytruda; MSD)[85] . However, the same predictive results have not been observed with nivolumab (Opdivo; Bristol-Myers Squibb). Despite the relative lack of success with this biomarker, for some patients the results are dramatic and can lead to immunotherapy supplanting all other treatments. At the time of writing, the FDA has expanded use of the approved Dako PD-L1 IHC 22C3 PharmDx assay (the CDx assay for pembrolizumab) from NSCLC to a variety of cancer types including gastric, gastroesophageal junction adenocarcinoma and cervical cancer[86] . However, in other tumour types (e.g. urological malignancies), PD-L1 over-expression does not appear to have any predictive ability for the benefit of immunotherapy. Findings from the pivotal Phase III KEYNOTE-189 study showed that using pembrolizumab in combination with the antimetabolite pemetrexed (Alimta; Eli Lilly) and a platinum-based agent such as cisplatin or carboplatin significantly improved overall survival, decreasing the risk of death by >50% compared with the use of chemotherapy alone[80]. In the UK, hospitals are currently providing initial triple therapy with platinum-based agents, pemetrexed and pembrolizumab to treat non-squamous NSCLC, followed by binary treatment with pembrolizumab and pemetrexed (as approved by the Cancer Drug Fund). Tumour mutational burden (TMB) and microsatellite instability-high (MSI-H) are new biomarkers that are being studied for use in personalised immunotherapy treatments[87] . While they are not yet approved as biomarkers for treatment selection, researchers are investigating the use of both to help clinicians accelerate their decisions in ruling in or out treatment options[88],[89] . For example, Bristol-Myers Squibb has a collaboration with Foundation Medicine Inc to use the FoundationOne CDx assay (Roche Products Ltd) to evaluate the use of TMB as a biomarker to assess the efficacy of a combination of two immune-oncology agents, nivolumab and ipilimumab (Yervoy , Bristol-Myers Squibb). The FoundationOne CDx assay, which was approved by the FDA in 2017, profiles MSI-H and TMB as well as 324 genes, and is approved to help identify best responders to 15 FDA-approved treatments including the anti-PD-1 immunotherapy agent pembrolizumab across five tumour types[88] . Results from the CheckMate-227 trial suggested that advanced NSCLC patients who are expressing high levels of TMB benefit from the nivolumab/ipilimumab combination, with higher progression-free survival observed in patients randomised to the combination arm compared with those given a single agent regardless of their PD-L1 status, suggesting that it may become a first-line therapy option in the future[87] . Bristol-Myers Squibb is currently using the FoundationOne CDx assay to evaluate the use of TMB as a predictive biomarker for nivolumab[89] . It should be noted that other more-general biomarkers such as vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR) have been identified as relevant to lung tumours in some studies[74],[90] .Metastatic colorectal cancer and other gastrointestinal tumours
The epithelial growth factor receptor (EGFR), a transmembrane ligand-induced receptor, is a predictive and prognostic biomarker over-expressed by tumour cells in around 70% of patients with mCRC[101] . Anti-EGFR monoclonal antibodies (mAbs), such as cetuximab and panitumumab, work by competitively inhibiting EGFR, preventing its interaction with endogenous ligands[101] with subsequent moderation of down-stream signaling pathways including RAS-RAF-MAPK and PI3K-AKT-mTOR[102] . In some cases, mutations in KRAS, NRAS and BRAF signaling proteins down-stream of EGFR may render colorectal cancers unresponsive to anti-EGFR treatment[101] . RAS genes are among the most frequently activated oncogenes in mCRC[101] . For example, mutations of Kristen RAS (KRAS) occur on exon 2, 3 and 4 in a ratio of 9:1[102] , and are considered to be useful predictive biomarkers for mCRC[101] . KRAS, NRAS and BRAF all predict resistance to anti-EGFR therapies and therefore highlighting their clinical benefit and importance as biomarkers[102],[103] . For example, in a study by Therkilsden et al., only 40–60% of patients with KRAS mutations responded to treatment[102] , Therefore, RAS mutation testing is now carried out to select the most likely responders to anti-EGFR treatment by identifying patients with mutations in signaling proteins including KRAS, BRAF and NRAS [101] . Laurent-Puig et al. have suggested that BRAF mutation status not only predicts the benefits of using anti-EGFR mABs, but also acts as an adverse prognostic biomarker for the course of mCRC[104] . KRAS mutations can be detected using a range of different techniques (see Table 3) including PCR (e.g. Cobas [Roche], Therascreen [Qiagen] and Idylla [Biocartis]), pyrosequencing, NGS (which allows the measurement of many mutations at once) or IHC, although the latter suffers from limited sensitivity and specificity[105] ,[106] . Human Epidermal Growth Factor Receptor 3 (HER3) over-expression has been established as a biomarker in patients harbouring the wild-type KRAS gene who are likely to benefit from anti-EGFR therapy such as panitumumab[107] . Results from the randomised PICCOLO trial revealed that patients with wild-type RAS together with increasing levels of HER3 expression, in addition to high levels of EGFR ligands such as epiregulin and amphiregulin, were associated with prolonged progression-free overall survival when given a combination of panitumumab and irinotecan compared with irinotecan alone [107],[108] . Although not presently tested for, in the future it may be possible to identify mCRC patients who may benefit from an anti-EGFR therapy based on the status of the HER3 and EGFR ligands together. The angiogenic biomarker vascular epithelial growth factor (VEGF) is associated with progression, invasion and metastasis of CRC[109] . It has been demonstrated that over-expression of VEGF mRNA in a primary tumour is highly correlated with a poor prognosis[109] . Therefore, this biomarker is potentially useful prognostically when diagnosing the stage of CRC[109] . In the same study it was found that hypoxia can activate the expression of VEGF in CRC tumour cells, thus promoting angiogenesis[110] . UDP-Glucuronosyltransferase 1A1 (UGT1A1) is an enzyme involved in the glucuronidation of SN-38, an active metabolite of irinotecan (Campto; Pfizer). It is a proven safety biomarker for this treatment in mCRC patients harbouring a polymorphism of the UGT1A1 gene (i.e. UGT1A1*28). This polymorphism is associated with a high risk of grade 3 or 4 haematological toxicity through decreased metabolism of SN-38. Thus, checking the UGT1A1 status of patients before initiating irinotecan therapy, and starting with a lower dose if the mutation is present, is recommended but not obligatory in many health systems globally. Testing is not put into practice by most oncologists[111],[112] , and the test is not routinely available in the NHS. PD-L1 expression associated with high microsatellite instability (MSI-H; Microsatellite Instability-High) or dMMR (Mismatch Repair Deficiency) biomarkers in CRC tumour cells can be used to identify patients who may benefit from immuno-oncology agents such as pembrolizumab or nivolumab[113] . Although pembrolizumab does not currently have an established role in the treatment of mCRC, in mid-2017 the FDA granted accelerated approval for the use of pembrolizumab in patients with unresectable or metastatic solid tumours with MSI-H or dMMR biomarkers. This was the first time that the FDA had accelerated a drug approval based on the genomic features of a tumour rather than its originating tissue or organ[114],[115] (see later). There are two clinically useful tests to detect MSI-H/dMMR biomarkers in cancer patents. The first involves the identification of MSI-H directly by molecular evaluation of poly-A microsatellites. The second is based on demonstration of lack of expression of MMR proteins through immunohistochemistry, which provides an indirect indication of an abnormal dMMR system[46] . The biomarkers, companion tests and associated drug treatments for mCRC are summarised in Table 3 below.Metastatic malignant melanoma
Around 50% of melanoma patients have BRAF mutations in their tumour cells[121] (see Figure 2)[122] . More than 90% are within codon 600 and, of these, more than 90% are a single nucleotide mutation in the kinase protein resulting in the change of a valine (V) at amino acid position 600 to a glutamic acid (E)[121] . This mutation, known asBRAF V600E, causes melanoma cells to proliferate aggressively. The first agent designed to target this mutated kinase, vemurafenib (Zelboraf; Roche), discovered by Plexxikon Inc but now owned by Daiichi-Sankyo/Genentech, proved very successful in the clinic and was approved by the FDA in 2011 for the treatment of late-stage melanoma. Another anti-BRAF V600E agent, dabrafenib (Tafinlar), followed for the treatment of advanced melanoma, and was initially approved by the FDA in 2013 as a single agent treatment for patients with BRAF V600E mutation-positive disease. Vemurafenib is significant in being the first drug to be discovered through a fragment-based lead discovery approach to gain regulatory approval. A PCR test for the BRAF V600E mutation is used to determine whether patients are suitable for these agents (see Supplementary PDF for table 4).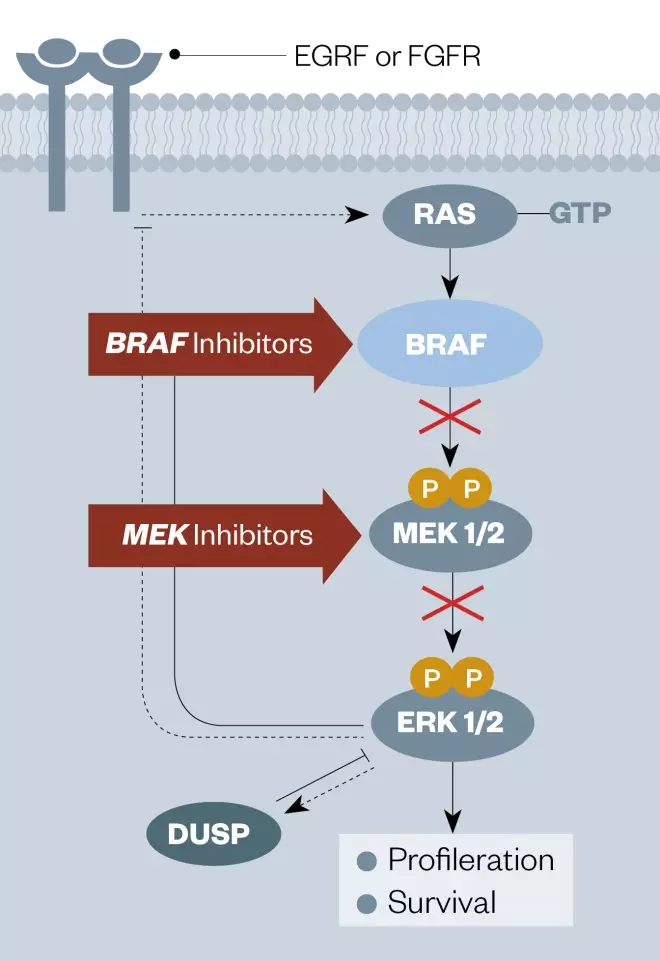
Figure 2: Diagram of the EGFR signaling pathway showing the points of intervention of BRAF and MEK kinase inhibitors
Source: Nat Rev Cancer[122]
EGFR: epidermal growth factor receptor FGFR: Fibroblast growth factor receptor GTP: Guanosine triphosphate MEK: MAPK/ERK kinase inhibitor DUSP: Dual specificity phosphatase ERK1/2: Extracellular-signal-regulated kinase 1/2
Ovarian cancer
Around 10% of invasive ovarian cancers have a hereditary basis, and many of these involve mutations in the BRCA1 and BRCA2 genes. Based on data from the Royal Marsden Foundation[41] , patients who carry a mutation in BRCA1 have a 40–60% chance of developing ovarian cancer, with the risk increasing after the age of 40. Those with BRCA2 mutations have a lower risk (i.e. 10–30%), although this also increases with age from the mid-late 40s. Patients with ovarian tumours containing BRCA1 and BRCA2 mutations tend to demonstrate higher platinum sensitivity over a longer period of time compared with those with no BRCA mutations. They also have an improved response to the PARP (Poly ADP Ribose Polymerase) inhibitors niraparib (Zejula; Tesaro UK Ltd), olaparib (Lynparza; AstraZeneca) and rucaparib (Rubraca; Clovis Oncology UK Ltd)[131],[132] . PARP inhibitors were developed through research on the different mechanisms of DNA single-strand break repair, through either homologous recombination or non-homologous end joining[133] . Inhibition of either one of these mechanisms separately has no effect on a tumour cell, whereas inhibition of both pathways simultaneously becomes a lethal event. Alongside Myriad Genetics’ BRCAnalysis CDx for olaparib and niraparib, a diagnostic test from Foundation Medicine has recently been approved by the FDA to identify patients suitable for treatment with rucaparib[134],[135] . More recently, AstraZeneca has been working with Myriad Genetics, using its MyChoice HRD Plus genetic test to select patients for treatment with a combination of olaparib and the anti-angiogenic agent bevacizumab (Avastin; Roche) (see Table 5)[136] . This test works by identifying and detecting patients with somatic mutations in their BRCA1/2 genes, as well as profiling 102 other genes[136] . It also identifies other mutagenic changes, such as loss of heterogeneity, telomeric allelic imbalance and large-scale state transitions[136] . Angiogenesis is a highly dynamic process regulated by pro- and anti-angiogenic modulating ligands, of which VEGF is one[137] . VEGF is considered to be the predominant growth factor expressed by tumour cells, and is regulated by internal factors such as hypoxia, acidosis, mechanical stress, and alterations in oncogenes and tumour suppression genes[137] . Over-expression of VEGF occurs in many solid tumour types, and is associated with shortened survival in ovarian cancer[137] . VEGF is expressed in more than 70% of ovarian cancers, and its level of expression is correlated with the stage of malignancy[137] . VEGFR-2 has been found in the tumour cells of 75% of ovarian cancer patients, and its inhibition is associated with a reduction in tumour growth and vascularisation in in vivo experiments, highlighting its importance in ovarian cancer prognosis and treatment[137] . The anti-angiogenic agent bevacizumab (Avastin), in combination with paclitaxel and carboplatin, has been approved by NICE for use in advanced disease as a first-line treatment[62],[126] . Other treatments for ovarian cancer include farletuzumab (MORAb-003), a monoclonal antibody that targets FR-alpha (a biomarker over-expressed in some cancers including ovarian cancer), which is presently being evaluated in combination with carboplatin and taxane in Phase III clinical trials in patients with relapsed platinum-sensitive ovarian cancer[138] . If successful, FR-alpha could be used in the future as a biomarker to select patients who may benefit from treatment with this agent. Clinically, ovarian cancer is usually diagnosed through an ultrasound in conjunction with measurement of the blood level of the CA125 protein, although diagnosis through tissue biopsy remains the gold standard, owing to the wide spectrum of characteristics of epithelial ovarian tumours. Morphotek Inc and Fujirebio Diagnostics are collaborating to develop, validate, manufacture and commercialise a diagnostic kit to detect CA125 II, a biomarker that is highly expressed in benign, borderline and malignant epithelial ovarian tumours[139] . On the basis of Phase III results, an anti-angiogenic gene therapy agent, ofranergene obadenovec (developed by Vascular Biogenics Ltd), was awarded Orphan Drug Case designation by the EMA in 2017[140] . This agent encodes a fusion protein that combines the extracellular and intra-membranal domains of human tumour necrosis factor 1 with a Fas receptor that contains a hypoxia-responsive element capable of driving cell death in the endothelium of blood vessels following expression of the chimeric protein[141] .Prostate cancer
Prostate cancer occurs most commonly in men aged >65 years, particularly in those of African descent[142] . It has also been established that African Americans with prostate cancer who express protein isoforms such as PIK3CD, FGFR3, TSC2 and RASGRP2 do not always respond to targeted therapies, potentially explaining the higher mortality rate in this population[143] . The best-known biomarker for prostate cancer is PSA (prostate-specific antigen), a protein produced by the prostate gland and typically over-expressed when the prostate becomes enlarged, inflamed or contains tumour cells[1] . PSA can be detected using a simple blood test, but has not yet been successfully targeted for the development of novel therapeutic agents. Instead, clinicians use PSA levels as part of a male health screen to trigger further investigations. Even though the FDA has approved the use of PSA testing as a screening tool for diagnosing prostate cancer, the test is non-specific because PSA levels can increase in both benign prostatic hyperplasia (BPH) and in prostate cancer, which leads to a high false-positive rate for prostate cancer[1] . Conversely, PSA expression can be low or normal in some cases of small cell prostatic carcinoma. At present, magnetic resonance imaging (MRI) is the gold standard for detecting prostatic carcinoma, which is then followed by a targeted biopsy if necessary. There are a number of treatment strategies and agents used to treat prostate cancer, but most are not directly linked to PSA levels. However, in early 2018 the FDA approved a novel agent apalutamide (Erleada ) for patients with non-metastatic prostate cancer resistant to standard hormone therapy (i.e. androgen deprivation therapy). In SPARTAN, a Phase III clinical trial for this agent that led to FDA approval, patients with castration-resistant prostate cancer but no metastatic disease were selected based on rapidly rising PSA levels (e.g. short PSA doubling time). They were randomly assigned to receive apalutamide or placebo in addition to ongoing androgen deprivation therapy, with a 70% reduction in mortality rate observed in the apalutamide treatment group. At present, patients are not specifically selected for treatment with apalutamide based on PSA levels, although research on this correlation is continuing. Mutated BRCA1 and BRCA2 genes are also becoming important biomarkers for prostate cancer, and have been used to select patients for therapy with PARP inhibitors such as olaparib[134] . In the TOPARP-A trial, blood samples from a small cohort of men with advanced prostate cancer were checked for DNA containing BRCA1 and BRCA2 mutations in the blood[131],[144] . The trial consisted of six patients who were selected to have homologous recombination deficiency (HRD) somatic mutation. The results suggested that HRD testing could be used to identify patients suitable for treatment with PARP inhibitors such as olaparib[134] . Other novel biomarkers linked to HRD have been found in 30% of patients with lethal prostate tumours[134] , although these have not yet been used for drug discovery purposes. Interestingly, studies have suggested that in prostate cancer patients carrying the BRCA2 mutation, the order in which some anticancer therapies are administered can affect progression-free survival. For example, the results of the PROREPAIR-B study (2013) demonstrated that administration of a taxane prior to an androgen signaling inhibitor led to inferior progression-free survival compared with non-carriers of BRCA2 [145] . However, reversing the order of administration led to a similar progression-free survival to non-carriers of the BRCA2 mutation[145] . Therefore, studies such as these suggest that genetic testing could be used to optimise treatment strategies for prostate cancer patients[145] . Phosphodiesterase-4D7 (PDE4D7) is an emerging prognostic biomarker for prostate cancer[146] . It was recently validated in a 503-patient study, which followed patients for 10 to 15 years to confirm the prognostic, and incremental value of PDE4D7 compared with established clinical risk metrics[146] . In 2018 MDxHealth Inc announced that it signed a worldwide licensing agreement with Philips for the rights to manufacture and market its tissue-based InformMDx test based on the PDE4D7 biomarker to stratify patients according to their risk of disease progression and the development of secondary tumours[146] . It is anticipated that this will provide useful information for clinicians to guide post-biopsy treatment decisions at the time of diagnosis, as well as treatment decisions following prostatectomy[146] . The main biomarkers used in prostate cancer, the tests used to identify them, and the use of this information in therapy are summarised in Table 6. Despite the gradual introduction of these PM approaches, the mainstay of treatment depending on the stage of the disease is presently based on androgen deprivation therapy, using agents such as luteinising hormone releasing hormone (LHRH) agonists (e.g. luserelin, goserelin, leuprorelin acetate or triptorelin), gonadotrophin-releasing hormone (GnRH) antagonists (e.g. degarelix), anti-androgen small molecule agents (e.g. bicalutamide, flutamide or cyproterone acetate) or traditional chemotherapy (e.g. docetaxel). Surgery, brachytherapy or radiotherapy are also important treatment strategies.Renal cell carcinoma
Von Hippel-Lindau (VHL) mutations are associated with many renal cell carcinoma (RCC) tumours resulting in higher levels of the heterodimeric transcription factor HIF1α (hypoxia inducible factor 1-α) and also VEGF (vascular endothelial growth Factor). VHL is associated with an autosomal dominant hereditary disorder, as well as being considered a tumour suppressor. The VHL protein targets the α-subunit of HIF1a and also associates with an E3 ligase, and has an important role in regulating hypoxia pathways both in tumour and healthy tissues[148],[149] , and growth factor-β signaling pathways in RCC[150] . Stabilisation of HIF1α corresponds to elevated VEGF levels together with increased vascularisation of VHL-associated tumours[148],[149] . A growing knowledge of the importance of VHL-HIF pathway in RCC has led to the development of treatment strategies involving multi-targeted tyrosine kinase inhibitors (e.g. pazopanib) and mTOR inhibitors (e.g. temsirolimus)[149] . VEGF is a potent promoter of tumour angiogenesis, and both mRNA and protein levels of VEGF are higher in RCC cells than in normal kidney cortex cells. In addition, VEGF and PDGF-B (platelet-derived growth factor B) expression have been found to be higher in papillary carcinoma cells than in other RCC subtypes[151] . Although not currently available in the NHS, the NexCourse Complete/Solid Genoptix-A Test has been used experimentally to measure over-expression of VEGFR 1-3 to select patients who may benefit from treatment with pazopanib (see Table 7). Some patients with RCC have been known to experience innate immune responses to their disease, which has led to exploration of the use of immunotherapies[152] . For example, the PD-L1 inhibitors nivolumab (Opdivo; Bristol-Myers Squibb Pharmaceuticals Ltc) and pembrolizumab (Keytruda; MSD) that inhibit the interaction between PD-L1 and PD-1 have been studied, but no CDx tests are presently available for these agents. Based on clinical studies, Weinstock et al have demonstrated that the PD-1/PD-L1 interaction is an important regulator of tumour immune tolerance and growth in RCC[152] , and that by blocking this interaction, progression of the disease can be slowed.Hepatocellular carcinoma
VEGF is a molecular biomarker that participates in processes such as the recruitment of endothelial cells through activation of the SAPK2/p38 MAP kinase module[153] , and the activation of receptors involved in the proliferation of tumour cells[154] . Studies have suggested that VEGF has an important role in the development, growth and metastasis of not just HCC, but in a number of other tumours types including lung, gastric, colorectal, and head and neck squamous cancers[154] . For HCC, the tyrosine kinase inhibitor sorafenib was developed to block synthesis of intercellular factors such as VEGF to regulate angiogenesis and thus the progression of the disease[154] . PD-L1 is a co-stimulatory ligand that is commonly expressed by HCC cells both in vitro and in vivo [155] . There is evidence to suggest that HCC patients whose tumours express PD-L1 are at higher risk of cancer reoccurrence. Furthermore, PD-L1 is a prognostic factor for tumour vascular invasion and encapsulation of HCC[155] . Overall, over-expression of PD-L1 in HCC correlates with tumour aggressiveness and an enhanced risk of post-operative recurrence[155] . Therefore, PD-L1 can be used in HCC as a biomarker for both prognosis and therapy with immune-oncology checkpoint inhibitors such as nivolumab (Opdivo; Bristol-Myers Squibb)[155] . The biomarkers used in hepatocellular carcinoma, the tests used to identify them, and the use of this information in therapy are summarised in Table 8.Papillary and medullary thyroid cancers
VEGF and PDGFR are commonly over-expressed in thyroid cancers[156] . For example, their presence correlates with an increase in vascularity and microvessel density in a papillary thyroid compared with that of a normal thyroid[156] . Cohen et al. have reported that VEGF and PDGFR over-expression are closely linked with tumour stage, size, nodal involvement, extra thyroidal invasion, distant metastasis and the risk of papillary thyroid cancer re-occurrence[148],[156] . By observing the multiple mutations within the BRAF gene, it has been established that its activation and, in turn, activation of the RAF/MEK/MAPK signaling pathway, is an important event in the development of papillary thyroid cancer[157] . The presence of the BRAF V600 mutation was found to be more prevalent in older age groups with lymph node and distant metastases, thus establishing it as an independent prognostic biomarker for recurrent and persistent disease[158] . The prognostic and diagnostic biomarkers for medullary thyroid cancer have been recently reviewed[159] . Oncogenic activation of BRAF contributes to the pathogenesis of several solid tumour types including hepatocellular carcinoma[160] . More information about the biomarkers and therapeutic agents associated with papillary and medullary thyroid cancer is summarised in Table 9.Pancreatic neuroendocrine tumours
Biopsy samples taken from pancreatic neuroendocrine tumours and pancreatic carcinomas frequently show over-expression of multiple biomarkers including PDGFR, C-kit (stem cell factor receptor) and VEGFR[161] . The kinase inhibitor sunitinib (Sutent; Pfizer) is capable of inhibiting these, and can delay the growth of pancreatic islet cell tumours by reducing endothelial cell density and the pericyte coverage of tumour vessels[161] . ERCC1 (excision repair cross-complementing gene-1) produces an excision nuclease within the nucleotide excision repair pathway. Based on IHC, over-expression of this gene has been shown to be a good predictive biomarker to guide initial treatment[162] . Research has suggested that over-expression of ERCC1 might be an effective predictor of response to FOLFIRINOX (a combination of folinic acid, fluorouracil, irinotecan and oxaliplatin) in metastatic pancreatic cancer[163] . An emerging area of interest is the identification of pancreatic tumours harbouring BRCA1/2 mutations that might be treated with PARP inhibitors such as olaparib (Lynparza) targeted to these mutations and other DNA damage repair defects. One study involving 3,315 metastatic pancreatic cancer patients (the POLO trial) utilised the Myriad Genetics BRACAnalysis CDx assay to identify suitable patients, and provided olaparib (Lynparza) to BRCA1/2 -positive individuals following 16 or more weeks of platinum-based chemotherapy[164] . This approach extended progression-free survival from 3.8 to 7.4 months compared with a placebo trial arm, although no significant health-related quality of life differences between the maintenance treatment and placebo arms was observed[163] . The investigators concluded that a strategic approach of first-line platinum-based chemotherapy followed by maintenance olaparib treatment could become a new standard of care for metastatic pancreatic cancer patients with germline BRCA1/2 mutations[163] . The biomarkers used in pancreatic neuroendocrine tumours the tests used to identify them, and the use of this information in therapy is summarised in Table 10.Bladder cancer
The checkpoint immune-oncology target PD-L1 has been a primary focus for the treatment of bladder cancer[165] , and it has been demonstrated that tumours expressing higher levels of PD-L1 are more likely to be assessed as high-grade, with greater frequencies of post-operative recurrence and poorer survival[165] . Durvalumab (Imfinzi; AstraZeneca), a monoclonal antibody targeted to PD-L1, was granted ‘breakthrough therapy’ designation by the FDA in 2016 for patients with PD-L1 positive inoperable or metastatic urothelial bladder cancer whose tumours have progressed during or after a standard platinum-based regimen[165] . This new agent is also being studied for its use in the treatment of NSCLC, hepatocellular carcinoma, mesothelioma, and head and neck, pancreatic and haematologic cancers[165] . Atezolizumab (Tecentriq) and Avelumab (Bavencio ) are also being studied for use in this disease setting. A new biomarker gaining relevance to the treatment of bladder cancer is the fibroblast growth factor receptor 2/3 (i.e. FGFR2 and FGFR3)[166] . In 2019, the FDA provided accelerated approval for erdafitinib (Balversa; Janssen Pharmaceuticals), which is targeted, to advanced adult bladder cancer patients with FGFR2 or FGFR3 mutations following treatment with platinum-based agents[166] . The FDA simultaneously approved the Therascreen FGFR RGQ RT-PCR Kit (Qiagen Inc) as a companion diagnostic test to select patients with FGFR3 or FGFR2 mutations who should benefit from erdafitinib therapy[166] . This was the first FDA approval of an FGFR-related therapeutic agent or companion diagnostic test[166] . As bladder tumours progress, they tend to spread into the muscularis propria surrounding the bladder. It has been shown that increased mutational frequency in genes that encode proteins involved in chromatin remodeling may prove useful in predicting whether a non-invasive tumour progresses to muscle invasion[167] . Two subtypes of non-invasive bladder cancer, GS1 and GS2, have been classified based on their genomic signatures, and in particular on an increase in KI67 labelling and up-regulation of mTORC1 signaling[168] . Interestingly, mutations in the histone demethylase enzyme-coding gene KDM6A are also thought to play a role, but are found predominantly in female patients[167] . These diagnostic and prognostic biomarkers appear to be key oncogenic drivers of the disease, and may prove to be useful therapeutic targets in the future[166],[167] . The main biomarker used in bladder cancer, the test used to identify it, and the use of this information in therapy is summarised in Table 11.Basal cell carcinoma
Basal cell carcinoma (BCC) is the most common type of non-melanoma skin cancer[169] . It is rarely treated with targeted therapeutic agents, the main treatment strategies being surgery, topical cytotoxic agents (e.g. 5-fluorouracil) and/or radiotherapy[168] . The occurrence of metastasis for BCC is very low (i.e. 0.0028–0.5500%) and, if diagnosed early, surgery is usually curative[168] . However, there has been an effort to develop targeted agents for patients in whom a BCC has recurred following surgery or radiation therapy, or who are not candidates for surgery or radiation therapy[170] . Hedgehog signaling is an important component of normal cellular development, regulating key target genes involved in modulation of the microenvironment (see Figure 3)[171],[172] . This pathway involves binding of the ligand SHH (‘sonic hedgehog’) to the PTCH1 (‘patched-1’) receptor[171] . In the absence of ligand, PTCH1 inhibits SMO (smoothened), a downstream protein in the pathway whose role is to migrate from the intracellular endosome to the cell membrane of the cilium where it activates the glioma-associated oncogene that stimulates expression of target genes[170] .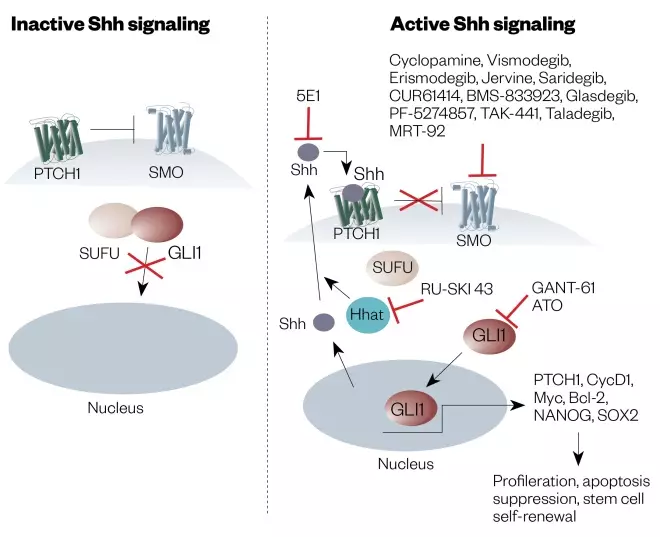
Figure 3: Diagram of the ‘Hedgehog signaling pathway’ and how to inhibit it
Source: Reproduced with permission from: Rimkuset al. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016;8(2):1-23
Shh: Sonic hedgehog SMO: Smoothened GLI: Glioma-associated oncogene homologue PTCH1: Patched1 SUFU: Suppressor of fused
Gastrointestinal stromal tumours
GISTs most commonly arise within the submucosa of the stomach and small bowel, with the cells of origin thought to be the pacemaker interstitial cells of Cajal. Diagnosis is supported with a positive immunohistochemistry for DOG1 (a calcium-dependent receptor-activated chloride channel) or C-Kit (also known as CD117) (see Table 13)[173],[174] . The majority of GISTs (i.e. around 85 to 90% of cases) have activating mutations in the C-Kit or PDGFRA receptor tyrosine kinases (TKIs). The C-kit mutations occur most frequently within exon 11, and affected tumours demonstrate a superior response to TKIs. The response of tumours with exon 9 mutations is less favourable, and those tumours with exon 17 mutations are usually resistant to first generation TKIs. Mutations in PDGFRA most commonly occur in exon 18, and affected tumours are also resistant to first generation TKIs[173],[174],[175] . Treatment with the first generation TKI imatinib (Glivec , Novartis) for three years is recommended as first-line adjuvant therapy for GISTs when there is a high risk of relapse owing to incomplete surgical excision or metastatic disease[173],[176] . Sunitinib (Sutent , Pfizer) is given when there are intolerable side effects with imatinib or tumour resistance develops[177] . Regorafenib (Stivarga , Bayer) is a second-generation multikinase inhibitor with FDA approval as an alternative therapy when there is intolerability to the other TKIs, or tumour resistance develops with both imatinib and sunitinib[174],[178] .Soft-tissue sarcomas
While rare, soft-tissue sarcomas (STSs) have an increasing incidence, although this currently remains under 1% of the UK population[180] . This heterogeneous group of malignant mesenchymal tumours has more than 50 subtypes, traditionally classified according to tissue differentiation that arise from a range of anatomical sites and demonstrate diverse clinical behaviour[181] . While the primary treatment is surgical resection or radiotherapy-based if resection is unachievable, tumour sensitivity to cytotoxic chemotherapies is also recognised in some subtypes[182],[183] . An understanding of the molecular biology of these tumours has led to the identification of diagnostic genetic aberrations. Around 20% of STS subtypes demonstrate an identifiable chromosomal translocation, of which around 66% result in fusion-protein activated pathways to which therapies may be targeted[184] . STS translocation aberrations and associated targets have been categorised according to fusion gene protein product function as previously reviewed along with suitable targeted therapeutic approaches[181],[184],[182],[183],[185],[186] . For example, PAX3-FOXO1 and PAX7-FOXO1 gene fusions are recurrently associated with morphologically distinctive alveolar rhabdomyosarcoma (ARMS), and are identifiable in 65% and 20% of cases respectively (184). ARMS most commonly presents in the group aged under 15 years, and the presence of a FOXO1 translocation is associated with a poorer prognosis. However, it has proven difficult to target chimeric transcription factor proteins directly[183], [184] .Similarly, Ewing sarcoma expresses prototypical transcription factor gene fusions such as EWSR1-FLI1 that is associated with down-stream over-expression of IGF1R (insulin growth factor 1 receptor 1)[185] . While Ewing sarcoma is typically chemotherapy sensitive (i.e. 70% of patients have five-year event-free survival in the non-metastatic group), recurrence is a poor prognostic scenario in which partial tumour response to the IGFR1 inhibitor trabectedin (Yondelis , Immedica) has been demonstrated[185] . Trabectedin has European (2007) and FDA (2015) approval for the treatment of metastatic liposarcoma and leiomyosarcoma, with other IGF1R inhibitors such as figitumumab and ganitumab providing objective responses as mono- or combination-therapies in early- and late-phase STS clinical trials respectively[183] . Other recurrent gene fusions occur in the protein kinases[184] , and TKIs have provided some overall survival benefit in subsets of STSs. For example, activity of the VEGF inhibitor pazopanib in non-adipocytic STS contributed toward FDA approval in 2012 for second-line use in this disease[182] . Sunitinib also has activity against VEGFR, PDGFR and RET, and cediranib has activity against multiple VEGFRs and KIT. Both of these TKIs are showing promise for the treatment of alveolar soft-part sarcomas in early-phase clinical trials[183] . In addition, mTOR inhibitors acting down-stream of PI3K/AKT in combination with TKIs or IGFR1 inhibitors have provided partial responses, but this is typically limited to a few individual STS patients in which general toxicity remains a major limitation[183] . Therefore, moving forward, there is a need to utilise biomarkers for the stratification of STS patients into those who are most likely to benefit from the increasing range of therapies available[183],[186] .Tumour agnostic agents and related biomarkers
Until recently, novel PM agents were designed (and approved) against cancer types originating in specific tissues within the body (e.g. breast, bowel and lung) based on particular genetic aberrations in biopsied cells (e.g. vemurafenib for BRAF V600E mutations in melanoma). However, there is a current trend to develop novel PM agents targeted towards biomarkers irrespective of which tumour or tissue types they arise in (i.e. so-called ‘tissue agnostic’ or ‘pan anticancer’ agents)[187],[188] . For example, in 2014 the FDA initially approved pembrolizumab (Keytruda , MSD) under the FDA Fast Track Development Program for use following treatment with ipilimumab (or after treatment with ipilimumab and a BRAF inhibitor) in advanced melanoma patients carrying a BRAF mutation[189] . However, in 2017 it was approved for use in non-small cell lung cancer, recurrent or metastatic head and neck cancer, refractory classical Hodgkin lymphoma, and urothelial carcinoma[190] . This agent works by targeting the cellular pathway involving PD-1/PD-L1, a receptor and ligand found on T-cells and some cancer cells respectively[191] . By blocking this pathway, pembrolizumab stimulates the body’s immune system to kill the tumour cells. Then, in 2017, the FDA granted accelerated approval for the use of pembrolizumab in the treatment of any adult or paediatric patients whose cancers have the specific genetic biomarkers of microsatellite instability-high (MSI-H) or mismatch repair deficiency (dMMR)[114],[192] . This was the first time that the FDA had approved a cancer treatment based on common biomarkers without regard for the tumour’s original location in the body[115],[189] . MSI-H and dMMR are genetic abnormalities that, if present, reduce the ability of cells to repair their DNA after mutation or other damage [193] . Tumours with these biomarkers are most commonly found in colorectal, endometrial and gastrointestinal cancers, but are also less commonly found in breast, prostate, bladder and thyroid cancers[113],[194] . Crucially, around 5% of patients with mCRC have MSI-H or dMMR biomarkers in their tumours[187],[193],[195] . Pembrolizumab was approved for this new indication using the accelerated approval pathway, through which the FDA may approve drugs for serious conditions where there is an unmet clinical need[114] . The safety and efficacy of pembrolizumab for this new approval were studied in patients carrying MSI-H or dMMR in their solid tumours enrolled in one of five uncontrolled, single-arm clinical trials. In some trials, patients were required to already have MSI-H or dMMR cancers prior to treatment, while in others a subgroup of patients were identified as having MSI-H or dMMR cancers by testing tumour samples after treatment had started. A total of 15 cancer types were identified among 149 patients enrolled across these five clinical trials, the most common being colorectal, endometrial and other gastrointestinal cancers. Of the 149 patients who received pembrolizumab in these trials, 39.6% had a complete or partial response, and for 78% of these patients, the response lasted six months or more[196] . In another example, researchers at Array BioPharma discovered larotrectinib (Vitrakvi ) (see Figure 4), an inhibitor of the Tropomyosin Kinase Receptors TrkA, TrkB and TrkC, which was subsequently licensed to Bayer and Loxo Oncology[196] . Larotrectinib was initially awarded orphan drug status by the FDA in 2015 for soft tissue sarcoma, which was followed by breakthrough therapy designation in 2016 for treatment of the approximately 1% of patients whose metastatic tumours are caused by chromosomal TRK gene fusions[196],[197],[198] . Clinical trial results were announced in 2017 and, as a result, the FDA approved larotrectinib in November 2018[199] .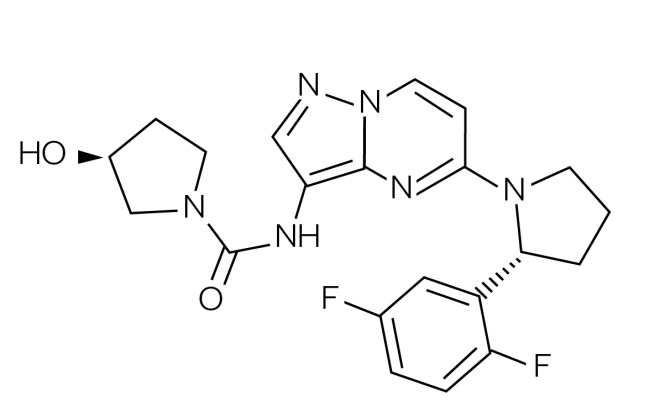
Figure 4: Structure of larotrectinib
Larotrectinib (Vitrakvi) is the first small molecule agent to be approved to treat any cancers containing certain mutations, rather than cancers of specific tissues (i.e. the approval was ‘tissue agnostic’)
Supportive therapies
PMs that target tumour cells while leaving healthy cells intact should, in principle, be devoid of side effects. Although this goal may be reached in the future, at present most, if not all, of the anticancer agents used in the clinic, whether classed as PMs or not, produce side effects in patients[202],[203] . Therefore, a number of supportive medicines are usually provided to treat these side effects to make the anticancer therapies more tolerable. They are essential for maintaining a good quality of life during treatment schedules, as well as ensuring that a patient completes their treatment without delays or dose reductions[204] . These supportive therapies are usually tailored to individual patients depending on which chemotherapy regimen they are receiving and which side effects they encounter, so in this context they are being used as part of a PM approach. Examples of supportive therapies include anti-emetics (e.g. ondansetron) to treat nausea and vomiting, and granulocyte-colony stimulating factors (GCSF) to reduce the risk of serious infection from myelosuppression[202],[203] . The majority of patients will be given anti-emetics before they receive chemotherapy and on the days following treatments, and a number of different agents may be tried before the best one for a particular patient is identified. If a PM agent is being used in combination with traditional anticancer regimens, other examples of supportive therapies include pyridoxine to counter the palmar-plantar erythrodysethesia resulting from fluorouracil or capecitabine treatment, folic acid to reduce the toxicity of pemetrexed or methotrexate, and omeprazole to protect the stomach from steroid-based treatments. The other major supportive medication is opioid therapy for pain associated with a tumour impacting on bones, nerves or other organs. Numerous opioid-based medications are available, and adjustments are made for individual patients to ensure that the best agent, dose and/or formulation are being used[205] .Funding of cancer drugs within the NHS
The growing number of new cancer therapies available, and their relatively high cost, places an increasing financial burden on the NHS and other healthcare systems globally. The new generations of PMs are particularly expensive owing to development costs and the fact that drug companies will sell fewer units owing to patients being selected for treatment (i.e. unlike the previous ‘block-buster’ model of drug sales where high demand reduced unit prices). Also, the use of PMs can be more expensive due to the need for CDx tests. Most observers agree that current funding models within the NHS are unsustainable, and that a major overhaul of how the system provides expensive anticancer agents to the entire UK population is required. At present, the provision of cancer drugs in England is a complicated process with multiple streams of funding. A number of anticancer drugs have been approved by NICE and are available through NHS England. In addition, a small number of anticancer medications are funded locally through clinical commissioning groups. In April 2011, the Cancer Drugs Fund (CDF) was introduced as a temporary solution to ensure that high-cost anticancer agents not routinely available through the NHS could be made available to patients. However, there was little control around the criteria for supplying drugs through the CDF, and the timeline for when drugs should exit the fund was not clear, which resulted in an increasing spend each year. It was agreed that the CDF was not a sustainable method for providing drugs to patients, and so a public consultation was conducted in 2015 by NHS England to establish a new model, with NICE overseeing the evaluation process, which was introduced in July 2016. In this new model, any new anticancer agents that are expected to receive marketing authorisation, or proposals for new indications for existing agents, are now referred to NICE for an initial appraisal. The advantage is that, as well as containing a clear exit strategy, the budget will not be allowed to overspend. However, if it does, the originating pharmaceutical company has a responsibility to contribute financially back into the CDF for their particular drug. At the point of appraisal by NICE, there are three potential outcomes. First, the new agent or new indication can be routinely commissioned, meaning that it will be funded by the CDF for 90 days. Following publication of the final NICE technology appraisal, it is then funded by NHS England. Second, the agent or new indication can be denied approval, meaning that it will only be available in exceptional circumstances through an individual funding request. Third, the new agent or indication can be entered into the interim CDF under a ‘managed access scheme’ to allow for more data to be collected. This option allows new agents or indications to be funded for a maximum of two years via the CDF, at which point a decision is made as to whether it will be routinely commissioned or not. Alternative access schemes can be either funded by the originating pharmaceutical company (i.e. through ‘compassionate access schemes’) or are evaluated via the MHRA through ‘early access to medicines’ schemes. Although these schemes are useful for providing new treatments to patients, they require a significant amount of administration and management resources.Funding of biomarker assays for cancer therapies
Although new PM therapies themselves will represent a significant cost to the NHS, the pharmacogenomic tests (i.e. CDxs) needed to support them will also add a significant cost. Most of the examples provided in this review are typically single biomarker assays, with a few having the potential to be carried out in parallel within the same assay (a process known as multiplexing). Eventually, multiplexing could reduce costs through, for example panel testing or whole exome sequencing. Overall, advances in NGS technologies have greatly enhanced the development and use of biomarker-driven cancer therapies. Genomic profiling has become significantly more cost-effective since the Human Genome Project, when the cost of sequencing the human genome was many millions of pounds and took more than a decade to complete. Current NGS platforms can sequence a patient’s genome in a few days at a cost of around £1,000. In particular, the affordability of, and improvements to, NGS have led to the commercialisation of NGS platforms that now have widespread use in both research and clinical decision-making. Despite the greater availability of tumour molecular profiling by NGS, the goal of improving patient outcomes while reducing overall costs (sometimes known as ‘value-based care’) remains challenging. A recent review by Gong et al. [35] has discussed the financial modelling of cost (efficiency) versus clinical benefit (effectiveness) and toxicity (safety) in relation to genomic profiling for cancer care, and it is clear that many health regimens around the world will find it challenging to deliver state-of-the-art genomics-based cancer care to all patients[31] . However, it is predicted that in the near future advances in NGS techniques with high-throughput functionality will enable the massive parallel sequencing of genomes at unprecedented rates (with targeted panels based on whole-exome or whole-genome sequencing), which should lower costs significantly.Conclusion
In the past three decades, the PM approach to cancer therapy has progressed from the concept phase to practical utility. This has significant implications for healthcare systems, such as the NHS, in that it creates the opportunity to provide anticancer agents to patients with a high certainty of providing clinical benefit[41] . However, these new therapies are expensive and require CDx tests to select patients. Although pharmaceutical companies are adjusting to this new paradigm by setting higher prices for novel PM-based anticancer therapies that sell in lower volume, government-funded healthcare organisations such as the NHS will come under increasing pressure to fund these new therapies because of their clinical effectiveness. For these new PM approaches, the growing burden on clinicians, pharmacists and other healthcare professionals will become increasingly significant[206] due to the additional requirements for identifying suitable patients using diagnostic tests followed by appropriate drug selection and variable dosing based on the test results. The complexity of this new treatment paradigm demands that healthcare professionals have not only the technical expertise and knowledge to deliver it, but also have the necessary communication skills to explain diagnostic test results and drug selection decisions clearly and confidently to patients and their carers, and other healthcare professionals. This review has provided an overview of the current status of the PM approach to cancer therapy with regard to the main types of solid tumours. A second part of this review, focusing on haematological malignancies, will also include a summary of the potential future applications of biomarkers in oncology. It should be noted that the accuracy and coverage of this review (and the one following on haematological malignancies) will be short-lived owing to the rapid advances being made in this area, with new PM therapies and biomarker assays entering the clinic at a remarkable rate. Each section of this review represents only a brief summary of what ideally should be a systematic review of thousands of publications. The reader should be aware of this, and is encouraged to look further into the literature in their area of interest.References
[1] Research Advocacy Network. Biomarkers in cancer: an introductory guide for advocates. 2010. Available at: http://www.researchadvocacy.org/sites/default/files/resources/BiomarkerinCancer_WebDownloadVersion.pdf (accessed November 2019)
[2] GenomeWeb. Brain Cancer Coalition Plans Precision Medicine Study. 2017. Available at: https://www.genomeweb.com/cancer/brain-cancer-coalition-plans-precision-medicine-study (accessed November 2019)
[3] Sciences AoM. Stratified, personalised or P4 medicine: a new direction for placing the patient at the centre of healthcare and health education. 2015. Available at: https://acmedsci.ac.uk/viewFile/564091e072d41.pdf (accessed November 2019)
[4] Robinson DR, Wu YM, Lonigro RJ et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548(7667):297–303. doi: 10.1038/nature23306
[5] Council NR. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. Washington, DC: The National Academies Press; 2011
[6] Collins FS & Varmus H. A new initiative on precision medicine. N Engl J Med 2015;372:793–795. doi: 10.1056/NEJMp1500523
[7] Coalition PM. Types of biomarkers. 2017. Available at: http://www.personalizedmedicinecoalition.org/Education/Types_of_Biomarkers (accessed November 2019)
[8] Kantarjian H, O’Brien S, Jabbour E et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: a single-institution historical experience. Blood 2012;119(9):1981–1987. doi: 10.1182/blood-2011-08-358135
[9] Prasad V. Perspective: the precision-oncology illusion. Nature 2016;537(7619):S63. doi: 10.1038/537S63a
[10] Engel J, Lategahn J & Rauh D. Hope and disappointment: covalent inhibitors to overcome drug resistance in non-small cell lung cancer. ACS Med Chem Lett 2016;7(1):2–5. doi: 10.1021/acsmedchemlett.5b00475
[11] Brandes AA, Franceschi E, Tosoni A et al. Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res 2008;14(4):957–960. doi: 10.1158/1078-0432.CCR-07-1810
[12] Sawyers CL. Opportunities and challenges in the development of kinase inhibitor therapy for cancer. Genes Dev 2003;17(24):2998–3010. doi: 10.1101/gad.1152403
[13] Matos LL, Trufelli DC, de Matos MG & da Silva Pinhal MA. Immunohistochemistry as an important tool in biomarkers detection and clinical practice. Biomark Insights 2010;5:9–20. doi: 10.4137/BMI.S2185
[14] Boots WebMD. FISH Test. 2017. Available at: https://www.webmd.com/cancer/fish-cancer-test#1 (accessed November 2019)
[15] Fluorescence in situ hybridization (FISH): an increasingly demanded tool for biomarker research and personalized medicine. Biomark Res 2014;2(1):3. doi: 10.1186/2050-7771-2-3
[16] Eastmond DA, Schuler M & Rupa DS. Advantages and limitations of using fluorescence in situ hybridization for the detection of aneuploidy in interphase human cells. Mutat Res 1995;348(4):153–162. doi: 10.1016/0165-7992(95)90003-9
[17] Meany DL, Sokoll LJ & Chan DW. Early detection of cancer: immunoassays for plasma tumor markers. Expert Opin Med Diagn 2009;3(6):597–605. doi: 10.1517/17530050903266830
[18] Bernard PS & Wittwer CT. Real-time PCR technology for cancer diagnostics. Clin Chem 2002;48(8):1178–1185. PMID: 12142370
[19] Kowalik A, Kowalewska M & Gozdz S. Current approaches for avoiding the limitations of circulating tumor cells detection methods – implications for diagnosis and treatment of patients with solid tumors. Transl Res 2017;185:58–84 e15. doi: 10.1016/j.trsl.2017.04.002
[20] Jahan-Tigh RR, Ryan C, Obermoser G & Schwarzenberger K. Flow cytometry. J Invest Dermatol 2012;132(10):1–6. doi: 10.1038/jid.2012.282
[21] Crutchfield CA, Thomas SN, Sokoll LJ & Chan DW. Advances in mass spectrometry-based clinical biomarker discovery. Clin Proteomics 2016;13(1):1. doi: 10.1186/s12014-015-9102-9
[22] Kamps R, Brandao RD, Bosch BJ et al. Next-generation sequencing in oncology: genetic diagnosis, risk prediction and cancer classification. Int J Mol Sci 2017;18(2)308. doi: 10.3390/ijms18020308
[23] Pharm J. DA issues guidance on developing new genetic tests for diagnosis and treatment of disease. 2018. Available at: https://www.pharmaceutical-journal.com/news-and-analysis/news-in-brief/fda-issues-guidance-on-developing-new-genetic-tests-for-diagnosis-and-treatment-of-disease/20204689.article (accessed November 2019)
[24] FDA. Use of public human genetic variant databases to support clinical validity for genetic and genomic-based in vitro diagnostics. 2018. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-public-human-genetic-variant-databases-support-clinical-validity-genetic-and-genomic-based-vitro (accessed November 2019)
[25] FDA. Considerations for design, development, and analytical validation of next generation sequencing (NGS) – based in vitro diagnostics (IVDs) intended to aid in the diagnosis of suspected germline diseases. 2018. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-design-development-and-analytical-validation-next-generation-sequencing-ngs-based (accessed November 2019)
[26] Terms NCIDoC. Biomarker. National Cancer Institute. 2019. Available at: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/biomarker (accessed November 2019)
[27] Mayeux R. Biomarkers: potential uses and limitations. NeuroRx 2004;1(2):182–188. doi: 10.1602/neurorx.1.2.182
[28] Drucker E & Krapfenbauer K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J 2013;4(1):7. doi: 10.1186/1878-5085-4-7
[29] Ray T. Q&A: Labceutics’ Jordan Clark on Industry, FDA Gap on Rx/Dx Codevelopment Principles. 2017. Available at: https://www.genomeweb.com/molecular-diagnostics/qa-labceutics-jordan-clark-industry-fda-gap-rxdx-codevelopment-principles (accessed November 2019)
[30] FDA. Companion diagnostics. 2017. Available at: https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm407297.htm (accessed November 2019)
[31] Scheerens H, Malong A, Bassett K et al. Current status of companion and complementary diagnostics: strategic considerations for development and launch. Clin Transl Sci 2017;10(2):84–92. doi: 10.1111/cts.12455
[32] Fridlyand J, Simon RM, Walrath JC et al. Considerations for the successful co-development of targeted cancer therapies and companion diagnostics. Nat Rev Drug Discov 2013;12(10):743–755. doi: 10.1038/nrd4101
[33] EUR-Lex. Directive 98/79/EC of the European Parliament and of the council of 27 October 1998 on in vitro diagnostic medical devices. OJ. 1998(1):1–37. Available at: http://data.europa.eu/eli/dir/1998/79/oj (accessed November 2019)
[34] Pharm J. EMA launches consultation on companion diagnostics and personalised medicine. Online: Pharm J. 2017. Available at: http://www.pharmaceutical-journal.com/news-and-analysis/news-in-brief/ema-launches-consultation-on-companion-diagnostics-and-personalised-medicine/20203318.article (accessed November 2019)
[35] Gong J, Pan K, Fakih M et al. Value-based genomics. Oncotarget 2018;9(21):15792–15815. doi: 10.18632/oncotarget.24353
[36] ASCO TAPUR. Targeted agent and profiling utilisation registry study, NCT02693535. 2017. Available at: https://www.tapur.org/researchers (accessed November 2019)
[37] Joenje H & Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2001;2(6):446–457. doi: 10.1038/35076590
[38] Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006;25(43):5875–5884. doi: 10.1038/sj.onc.1209878
[39] Konicek BW, Capen AR, Credille KM et al. Merestinib (LY2801653) inhibits neurotrophic receptor kinase (NTRK) and suppresses growth of NTRK fusion bearing tumors. Oncotarget 2018;9(17):13796–13806. doi: 10.18632/oncotarget.24488
[40] Jamieson AM, Yu S, Annicelli CH & Medzhitov R. Influenza virus-induced glucocorticoids compromise innate host defense against a secondary bacterial infection. Cell Host Microbe 2010;7(2):103–114. doi: 10.1016/j.chom.2010.01.010
[41] The Royal Marsden NHS Foundation Trust. A beginner’s guide to BRCA1 and BRCA2. 2016. Available at: https://www.royalmarsden.nhs.uk/sites/default/files/files_trust/beginners-guide-to-brca1-and-brca2.PDF (accessed November 2019)
[42] National Cancer Institute. Solid tumor. 2003. Available at: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/solid-tumor (accessed November 2019)
[43] National Cancer Institute. Cancer classification. Available at: https://training.seer.cancer.gov/disease/categories/classification.html (accessed November 2019)
[44] Swanton C & Caldas C. Molecular classification of solid tumours: towards pathway-driven therapeutics. Br J Cancer 2009;100(10):1517–1522. doi: 10.1038/sj.bjc.6605031
[45] Rakha EA, Reis-Filho JS, Baehner F et al. Breast cancer prognostic classification in the molecular era: the role of histological grade. Breast Cancer Res 2010;12(4):207. doi: 10.1186/bcr2607
[46] Sawyer E, Roylance R, Petridis C et al. Genetic predisposition to in situ and invasive lobular carcinoma of the breast. PLoS Genet 2014;10(4):e1004285. doi: 10.1371/journal.pgen.1004285
[47] Dai X, Xiang L, Li T & Bai Z. Cancer hallmarks, biomarkers and breast cancer molecular subtypes. J Cancer 2016;7(10):1281–1294. doi: 10.7150/jca.13141
[48] Carlson RW, Moench SJ, Hammond ME et al. HER2 testing in breast cancer: NCCN Task Force report and recommendations. J Natl Compr Cancer Netw 2006;4(Suppl 3):S1–22; quiz S3–4. PMID: 16813731
[49] Mohammed H, Russell IA, Stark R et al. Progesterone receptor modulates ERα action in breast cancer. Nature 2015;523:313. doi: 10.1038/nature14583
[50] National Institute of Health and Care Excellence. Do not do recommendation. 2009. Available at: https://www.nice.org.uk/donotdo/do-not-routinely-assess-progesterone-receptor-status-of-tumours-in-patients-with-invasive-breast-cancer (accessed November 2019)
[51] Campbell EJ, Tesson M, Doogan F et al. The combined endocrine receptor in breast cancer, a novel approach to traditional hormone receptor interpretation and a better discriminator of outcome than ER and PR alone. Br J Cancer 2016;115(8):967–973. doi: 10.1038/bjc.2016.206
[52] National Cancer Institute. HER2’s genetic link to breast cancer spurs development of new treatments. Available at: https://www.cancer.gov/research/progress/discovery/HER2 (accessed November 2019)
[53] Rhodes A, Jasani B, Balaton AJ et al. Frequency of oestrogen and progesterone receptor positivity by immunohistochemical analysis in 7016 breast carcinomas: correlation with patient age, assay sensitivity, threshold value, and mammographic screening. J Clin Pathol 2000;53(9):688–696. doi: 10.1136/jcp.53.9.688
[54] Rakha EA, Pinder SE, Bartlett JM et al. Updated UK recommendations for HER2 assessment in breast cancer. J Clin Pathol 2015;68(2):93–99. doi: 10.1136/jclinpath-2014-202571
[55] Bartlett JM, Starczynski J, Atkey N et al. HER2 testing in the UK: recommendations for breast and gastric in-situ hybridisation methods. J Clin Pathol 2011;64(8):649–653. doi: 10.1136/jcp.2011.089847
[56] The Royal College of Pathologists. Guidelines for non-operative diagnostic procedures and reporting in breast cancer screening. 2016. Available at: https://www.rcpath.org/uploads/assets/4b16f19c-f7bd-456c-b212f557f8040f66/G150-Non-op-reporting-breast-cancer-screening-Feb17.pdf (accessed November 2019)
[57] Meijers-Heijboer H, van den Ouweland A, Klijn J et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations.Nat Genet 2002;31(1):55–59. doi: 10.1038/ng879
[58] Komen S G. BRCA1 and BRCA2. 2017. Available at: https://ww5.komen.org/BreastCancer/BRCA1andBRCA2.html (accessed November 2019)
[59] Yoshida K & Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci 2005;95(11):866–871. doi: 10.1111/j.1349-7006.2004.tb02195.x
[60] Copson ER, Maishman TC, Tapper WJ et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study. Lancet Onc 2018;19(2):169–180. doi: 10.1016/S1470-2045(17)30891-4
[61] Yu Q, Sicinska E, Geng Y et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006;9(1):23–32. doi: 10.1016/j.ccr.2005.12.012
[62] Turner NC, Ro J, Andre F et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med 2015;373(3):209–219. doi: 10.1056/NEJMoa1505270
[63] Ray T. Novartis drug shows promise in PIK3CA mutated breast cancer, potential utility of ctDNA testing. 2018. Available at: h ttps://www.genomeweb.com/cancer/novartis-drug-shows-promise-pik3ca-mutated-breast-cancer-potential-utility-ctdna-testing (accessed November 2019)
[64] GenomeWeb. FDA approves Qiagen’s PIK3CA test as companion test for Novartis breast cancer drug. 2019. Available at: https://www.genomeweb.com/regulatory-news/fda-approves-qiagens-pik3ca-test-companion-test-novartis-breast-cancer-drug (accessed November 2019)
[65] Astor L. Only modest benefit seen with taselisib in PIK3CA-mutant breast cancer. Online: OncLive. 2018. Available at: https://www.onclive.com/conference-coverage/asco-2018/only-modest-benefit-seen-with-taselisib-in-pik3camutant-breast-cancer (accessed November 2019)
[66] GenomeWeb. UK’s NHS adopts NICE advice to provide oncotype DX for intermediate risk breast cancer patients. 2015. Available at: https://www.genomeweb.com/cancer/uks-nhs-adopts-nice-advice-provide-oncotype-dx-intermediate-risk-breast-cancer-patients (accessed November 2019)
[67] GenomeWeb. NICE draft guidance reverses endorsement of oncotype Dx to guide chemotherapy. 2018. Available at: https://www.genomeweb.com/molecular-diagnostics/nice-draft-guidance-reverses-endorsement-oncotype-dx-guide-chemotherapy (accessed November 2019)
[68] GenomeWeb. Myriad genetics submits supplementary PMA for BRACanalysis CDx. 2018. Available at: https://www.genomeweb.com/molecular-diagnostics/myriad-genetics-submits-supplementary-pma-bracanalysis-cdx (accessed November 2019)
[69] GenomeWeb. FDA accepts myriad BRACanalysis CDx supplementary PMA for lynparza in breast cancer indication. 2017. Available at: https://www.genomeweb.com/regulatory-news/fda-accepts-myriad-bracanalysis-cdx-supplementary-pma-lynparza-breast-cancer (accessed November 2019)
[70] GenomeWeb. FDA approves myriad genetics BRCA CDx with Pfizer’s metastatic breast cancer drug. 2018. Available at: https://www.genomeweb.com/regulatory-news/fda-approves-myriad-genetics-brca-cdx-pfizers-metastatic-breast-cancer-drug (accessed November 2019)
[71] National Institute for Health and Care Excellence. Guidance and advice list. 2017. Available at: https://www.nice.org.uk/guidance/published?type=ta
[72] Viapath. Test index. 2014. Available at: http://www.viapath.co.uk/tests-index (accessed November 2019)
[73] Viapath. New tests. 2013. Available at: http://www.viapath.co.uk/new-tests (accessed November 2019)
[74] Lemjabbar-Alaoui H, Hassan OU, Yang YW et al. Lung cancer: biology and treatment options. Biochim Biophys Acta 2015;1856(2):189–210. doi: 10.1016/j.bbcan.2015.08.002
[75] Riely GJ. The use of first-generation tyrosine kinase inhibitors in patients with NSCLC and somatic EGFR mutations. Lung Cancer 2008;60(2):S19–S22. doi: 10.1016/S0169-5002(08)70101-6
[76] Remon J, Caramella C, Jovelet C et al. Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Ann Oncol 2017;28(4):784–790. doi: 10.1093/annonc/mdx017
[77] Liao BC, Lin CC & Yang JC. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr Opin Oncol 2015;27(2):94–101. doi: 10.1097/CCO.0000000000000164
[78] Korpanty GJ, Graham DM, Vincent MD et al. Biomarkers that currently affect clinical practice in lung cancer: EGFR, ALK, MET, ROS-1, and KRAS. Front Oncol 2014;4:204. doi: 10.3389/fonc.2014.00204
[79] Raica M & Cimpean AM. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals (Basel). 2010;3(3):572–599. doi: 10.3390/ph3030572
[80] Berger W, Setinek U, Mohr T et al. Evidence for a role of FGF-2 and FGF receptors in the proliferation of non-small cell lung cancer cells. Int J Cancer 1999;83(3):415–423. doi: 10.1002/(SICI)1097-0215(19991029)83:3<415::AID-IJC19>3.0.CO;2-Y
[81] Kono SA, Marshall ME, Ware KE & Heasley LE. The fibroblast growth factor receptor signaling pathway as a mediator of intrinsic resistance to EGFR-specific tyrosine kinase inhibitors in non-small cell lung cancer. Drug Resist Updat 2009;12(4–5):95–102. doi: 10.1016/j.drup.2009.05.001
[82] Planchard D, Smit EF, Groen HJM et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 2017;18(10):1307–1316. doi: 10.1016/S1470-2045(17)30679-4
[83] Harris J. FDA approves dabrafenib/trametinib combo for BRAF+ NSCLC. Online: OncLive. 2017. Available at: http://www.onclive.com/web-exclusives/fda-approves-dabrafenibtrametinib-combo-for-braf-nsclc (accessed November 2019)
[84] DiBardino DM, Rawson DW, Saqi A et al. Next-generation sequencing of non-small cell lung cancer using a customized, targeted sequencing panel: emphasis on small biopsy and cytology. Cytojournal 2017;14:7. doi: 10.4103/1742-6413.202602
[85] Evans B & Evans S. Immune checkpoint inhibitors in cancer: pharmacology and toxicities. Pharm J 2018;300(7913):10. doi: 10.1211/pj.2018.20204831
[86] GenomeWeb. FDA approves expanded use of agilent assay as Keytruda CDx for cervical cancer. 2018. Available at: https://www.genomeweb.com/molecular-diagnostics/fda-approves-expanded-use-agilent-assay-keytruda-cdx-cervical-cancer (accessed November 2019)
[87] Ray T. BMS immunotherapy combo study backs FoundationOne CDx tumor mutational burden indication. 2018. Available at: https://www.genomeweb.com/molecular-diagnostics/bms-immunotherapy-combo-study-backs-foundationone-cdx-tumor-mutational-burden (accessed November 2019)
[88] GenomeWeb. Merck, Foundation Medicine to develop companion diagnostics for immunotherapy. 2018. Available at: https://www.genomeweb.com/cancer/merck-foundation-medicine-develop-companion-diagnostics-immunotherapy (accessed November 2019)
[89] Karow J. Two initiatives seek to harmonize tumor mutational burden testing. 2018. Available at: https://www.genomeweb.com/cancer/two-initiatives-seek-harmonize-tumor-mutational-burden-testing (accessed November 2019)
[90] Noskovicova N, Petrek M, Eickelberg O & Heinzelmann K. Platelet-derived growth factor signaling in the lung. From lung development and disease to clinical studies. Am J Resp Cell Mol Biol 2015;52(3):263–284. doi: 10.1165/rcmb.2014-0294TR
[91] GenomeWeb. Cancer genetics gets NY state approval for Thermo Fisher lung cancer CDx. 2017. Available at: https://www.genomeweb.com/molecular-diagnostics/cancer-genetics-gets-ny-state-approval-thermo-fisher-lung-cancer-cdx (accessed November 2019)
[92] Ray T. After approval of next-gen sequencing CDx panel, Thermo Fisher seeks rapid expansion of indications. 2017. Available at: https://www.genomeweb.com/clinical-sequencing/after-approval-next-gen-sequencing-cdx-panel-thermo-fisher-seeks-rapid-expansion (accessed November 2019)
[93] GenomeWeb. Quest to offer Thermo Fisher’s Oncomine Dx target test. 2018. Available at: https://www.genomeweb.com/clinical-lab-management/quest-offer-thermo-fishers-oncomine-dx-target-test (accessed November 2019)
[94] GenomeWeb. Qiagen gets FDA OK for Eepanded use of EGFR CDx for lung cancer drug. 2018. Available at: https://www.genomeweb.com/pcr/qiagen-gets-fda-ok-expanded-use-egfr-cdx-lung-cancer-drug (accessed November 2019)
[95] GenomeWeb. FDA approves expanded use of Qiagen EGFR CDx in lung cancer. 2018. Available at: https://www.genomeweb.com/regulatory-news/fda-approves-expanded-use-qiagen-egfr-cdx-lung-cancer (accessed November 2019)
[96] GenomeWeb. Thermo Fisher inks new pharma partnerships to expand CDx use of Oncomine NGS test. Online: GenomeWeb. 2018. Available at: https://www.genomeweb.com/molecular-diagnostics/thermo-fisher-inks-new-pharma-partnerships-expand-cdx-use-oncomine-ngs-test (accessed November 2019)
[97] HTG EdgeSeq ALKPlus assay EU for ALK status testing in non-small-cell lung cancer. Medtech innovation briefing [MIB128]. 2017. Available at: https://www.nice.org.uk/advice/mib128 (accessed November 2019)
[98] FoundationOne CDx. FoundationOne CDxTM Technical Information. 2017. Available at: https://www.accessdata.fda.gov/cdrh_docs/pdf17/P170019C.pdf (accessed November 2019)
[99] Ashford M. With promising data in early-stage NSCLC, PGDx continues TMB test kit development. 2018. Available at: https://www.genomeweb.com/cancer/promising-data-early-stage-nsclc-pgdx-continues-tmb-test-kit-development (accessed November 2019)
[100] Ashford M. With promising data in early-stage NSCLC, PGDx continues TMB test kit development. 2018. Available at: https://www.genomeweb.com/cancer/promising-data-early-stage-nsclc-pgdx-continues-tmb-test-kit-development (accessed November 2019)
[101] Kalia M. Biomarkers of colorectal cancer. J Cancer Biol Res 2015;3(1):1058
[102] Therkildsen C, Bergmann TK, Henrichsen-Schnack T et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol 2014;53(7):852–864. doi: 10.3109/0284186X.2014.895036
[103] De Roock W, De Vriendt V, Normanno N et al. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol 2011;12(6):594-603. doi: 10.1016/S1470-2045(10)70209-6
[104] Laurent-Puig P, Cayre A, Manceau G et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 2009;27(35):5924–5930. doi: 10.1200/JCO.2008.21.6796
[105] Cree IA. Diagnostic RAS mutation analysis by polymerase chain reaction (PCR). Biomol Detect Quantif 2016;8:29–32. doi: 10.1016/j.bdq.2016.05.001
[106] Cree IA. Progress and potential of RAS mutation detection for diagnostics and companion diagnostics. Expert Rev Mol Diagn 2016;16(10):1067–1072. doi: 10.1080/14737159.2016.1221345
[107] GenomeWeb. High HER3 levels identify colorectal cancer patients who will respond to Anti-EGFR therapy. 2017. Available at: https://www.genomeweb.com/gene-expression-research/high-her3-levels-identify-colorectal-cancer-patients-who-will-respond-anti (accessed November 2019)
[108] Seligmann JF, Hatch AJ, Richman SD et al. Association of tumor HER3 messenger RNA expression with panitumumab efficacy in advanced colorectal cancer. JAMA Oncol 2017. doi: 10.1001/jamaoncol.2017.3168
[109] Ishigami SI, Arii S, Furutani M et al. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br J Cancer 1998;78(10):1379–1384. doi: 10.1038/bjc.1998.688
[110] Cao D, Hou M, Guan YS et al. Expression of HIF-1alpha and VEGF in colorectal cancer: association with clinical outcomes and prognostic implications. BMC Cancer 2009;9(432):432. doi: 10.1186/1471-2407-9-432
[111] Iyer L, Das S, Janisch L et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–47. doi: 10.1038/sj/tpj/6500072
[112] Toffoli G, Cecchin E, Corona G et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol 2006;24(19):3061–3068. doi: 10.1200/JCO.2005.05.5400
[113] Rosenbaum MW, Bledsoe JR, Morales-Oyarvide V et al. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod Pathol 2016;29(9):1104–1112. doi: 10.1038/modpathol.2016.95
[114] Broderick JM. FDA approves pembrolizumab for microsatellite instability-high and mismatch repair deficient cancers. 2017. Available at: http://www.onclive.com/web-exclusives/fda-approves-pembrolizumab-for-microsatellite-instabilityhigh-and-mismatch-repair-deficient-cancers (accessed November 2019)
[115] GenomeWeb. FDA approves first drug for tumor’s biomarker indication. 2017. Available at: https://www.genomeweb.com/molecular-diagnostics/fda-approves-first-drug-tumors-biomarker-indication (accessed November 2019)
[116] GenomeWeb. Biocartis, Amgen sign CDx development deal for Idylla RAS biomarker tests. 2017. Available at: https://www.genomeweb.com/business-news/biocartis-amgen-sign-cdx-development-deal-idylla-ras-biomarker-tests (accessed November 2019)
[117] GenomeWeb. Biocartis gets CE marking for two colorectal cancer liquid biopsy tests. 2017. Available at: https://www.genomeweb.com/pcr/biocartis-gets-ce-marking-two-colorectal-cancer-liquid-biopsy-tests (accessed November 2019)
[118] GenomeWeb. Colorectal cancer clues provided by Polyp Profiles. 2018. Available at: https://www.genomeweb.com/sequencing/colorectal-cancer-clues-provided-polyp-profiles (accessed November 2019)
[119] Druliner BR, Wang P, Bae T et al. Molecular characterization of colorectal adenomas with and without malignancy reveals distinguishing genome, transcriptome and methylome alterations. Sci Rep 2018;8(1):3161. doi: 10.1038/s41598-018-21525-4
[120] Gilmore J. PavMed spinoff lucid diagnostics aims to market molecular Barrett’s esophagus test in 2019. 2018. Available at: https://www.genomeweb.com/sequencing/pavmed-spinoff-lucid-diagnostics-aims-market-molecular-barretts-esophagus-test-2019 (accessed November 2019)
[121] Ascierto PA, Kirkwood JM, Grob JJ et al. The role of BRAF V600 mutation in melanoma. J Transl Med 2012;10:85. doi: 10.1186/1479-5876-10-85
[122] Caunt CJ, Sale MJ, Smith PD & Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer 2015;15(10):577–592. doi: 10.1038/nrc4000
[123] Pires da Silva I, Wang KYX, Wilmott JS et al. Distinct molecular profiles and immunotherapy treatment cutcomes of V600E and V600K BRAF-mutant melanoma. Clin Cancer Res 2019;25(4). doi: 10.1158/1078-0432.CCR-18-1680
[124] Grob JJ, Amonkar MM, Karaszewska B et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncol 2015;16(13):1389–1398. doi: 10.1016/S1470-2045(15)00087-X
[125] Long GV, Stroyakovskiy D, Gogas H et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015;386(9992):444–451. doi: 10.1016/S0140-6736(15)60898-4
[126] Buchbinder EI & Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol 2016;39(1):98–106. doi: 10.1097/COC.0000000000000239
[127] Inc B. Technical sheet Idyllaâ„¢ BRAF Mutation Test. 2016. Available at: https://www.biocartis.com/sites/default/files/130387-TechSheet-BRAF-A4_web.pdf (accessed November 2019)
[128] Reiman A, Kikuchi H, Scocchia D et al. Validation of an NGS mutation detection panel for melanoma. BMC Cancer 2017;17(1):150. doi: 10.1186/s12885-017-3149-0
[129] Tops BB, Normanno N, Kurth H et al. Development of a semi-conductor sequencing-based panel for genotyping of colon and lung cancer by the Onconetwork consortium. BMC Cancer 2015;15(26):26. doi: 10.1186/s12885-015-1015-5
[130] Kikuchi H, Reiman A, Nyoni J et al. Development and validation of a TaqMan Array for cancer mutation analysis. Pathogenesis 2016;3(1):1–8. doi: 10.1016/j.pathog.2016.02.001
[131] Goodall J, Mateo J, Yuan W et al. Circulating free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov 2017:0F1–0F13. doi: 10.1158/2159-8290.CD-17-0261
[132] Lakhani SR, Manek S, Penault-Llorca F et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin Cancer Res 2004;10:2473–2481. doi: 10.1158/1078-0432.CCR-1029-3
[133] Meehan RS & Chen AP. New treatment option for ovarian cancer: PARP inhibitors. Gynecol Oncol Res Pract 2016;3(3):3. doi: 10.1186/s40661-016-0024-7
[134] Ashford M. Liquid biopsy may guide PARP inhibitor therapy for prostate cancer. 2017. Available at: https://www.genomeweb.com/cancer/liquid-biopsy-may-guide-parp-inhibitor-therapy-prostate-cancer (accessed November 2019)
[135] Ray T. Foundation Medicine’s regulatory win for NGS CDx sets precedent; adoption challenge remains. 2016. Available at: https://www.genomeweb.com/molecular-diagnostics/foundation-medicines-regulatory-win-ngs-cdx-sets-precedent-adoption-challenge (accessed November 2019)
[136] GenomeWeb. AstraZeneca, Myriad Ink Rx/Dx collaboration for MyChoice HRD Plus. 2018. Available at: https://www.genomeweb.com/molecular-diagnostics/astrazeneca-myriad-ink-rxdx-collaboration-mychoice-hrd-plus (accessed November 2019)
[137] Spannuth WA, Nick AM, Jennings NB et al. Functional significance of VEGFR-2 on ovarian cancer cells. Int J Cancer 2009;124(5):1045–1053. doi: 10.1002/ijc.24028
[138] GenomeWeb. Morphotek, Fujirebio to develop CA125 II assay as CDx. 2017. Available at: https://www.genomeweb.com/business-news/morphotek-fujirebio-develop-ca125-ii-assay-cdx (accessed November 2019)
[139] Heger M. Dana-Farber team developing MicroRNA liquid biopsy to diagnose ovarian cancer. 2017. Available at: https://www.genomeweb.com/molecular-diagnostics/dana-farber-team-developing-microrna-liquid-biopsy-diagnose-ovarian-cancer (accessed November 2019)
[140] Middleman M. VBL Therapeutics announces orphan drug designation for VB-111 in Europe. 2017. Available at: https://www.biospace.com/article/releases/vbl-therapeutics-announces-orphan-drug-designation-for-vb-111-in-europe (accessed November 2019)
[141] Triozzi PL & Borden EC. VB-111 for cancer. Expert Opin Biol Ther 2011;11(12):1669–1676. doi: 10.1517/14712598.2011.618122
[142] Society AC. Prostate cancer. In: Society AC, editor: American Cancer Society; 2016. 1–22. Available at: https://www.cancer.org/content/dam/cancer-org/cancer-control/en/presentations/prostate-cancer-presentation-short-version.pdf (accessed November 2019)
[143] GenomeWeb. Prostate cancers from African Americans show splice variants implicated in tumor invasiveness. 2017. Available at: https://www.genomeweb.com/microarrays-multiplexing/prostate-cancers-african-americans-show-splice-variants-implicated-tumor (accessed November 2019)
[144] Institute of Cancer Research. A phase II trial of olaparib in patients with advanced castration resistant prostate cancer (TOPARP). 2012. Available at: https://clinicaltrials.gov/ct2/show/NCT01682772 (accessed November 2019)
[145] GenomeWeb. BRCA2 mutations linked to poor treatment response, outcomes in prostate cancer. 2019. Available at: https://www.genomeweb.com/cancer/brca2-mutations-linked-poor-treatment-response-outcomes-prostate-cancer (accessed November 2019)
[146] GenomeWeb. MDxHealth licenses prognostic prostate cancer biomarker from Philips. 2018. Available at: https://www.genomeweb.com/business-news/mdxhealth-licenses-prognostic-prostate-cancer-biomarker-philips (accessed November 2019)
[147] NHS Choices. PSA testing: Prostate cancer. 2015. Available at: https://www.nhs.uk/conditions/prostate-cancer/psa-testing (accessed November 2019)
[148] Maranchie JK, Vasselli JR, Riss J et al. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 2002;1(3):247–255. doi: 10.1016/S1535-6108(02)00044-2
[149] Cowey CL & Rathmell WK. VHL gene mutations in renal cell carcinoma: role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep 2009;11(2):94–101. doi: 10.1007/s11912-009-0015-5
[150] Mallikarjuna P, Sitaram RT, Landstrom M et al. VHL status regulates transforming growth factor-beta signaling pathways in renal cell carcinoma. Oncotarget 2018;9(23):16297–16310. doi: 10.18632/oncotarget.24631
[151] Song SH, Jeong IG, You D et al. VEGF/VEGFR2 and PDGF-B/PDGFR-beta expression in non-metastatic renal cell carcinoma: a retrospective study in 1,091 consecutive patients. Int J Clin Exp Pathol 2014;7(11):7681–7689. PMID: 25550804
[152] Weinstock M & McDermott D. Targeting PD-1/PD-L1 in the treatment of metastatic renal cell carcinoma. Ther Adv Urol 2015;7(6):365–377. doi: 10.1177/1756287215597647
[153] Lamalice L, Houle F & Huot J. Phosphorylation of Tyr1214 within VEGFR-2 triggers the recruitment of Nck and activation of Fyn leading to SAPK2/p38 activation and endothelial cell migration in response to VEGF. J Biol Chem 2006;281(45):34009–34020. doi: 10.1074/jbc.M603928200
[154] Cao G, Li X, Qin C & Li J. Prognostic value of VEGF in hepatocellular carcinoma patients treated with sorafenib: a meta-analysis. Med Sci Monit 2015;21:3144–3151. doi: 10.12659/msm.894617
[155] Gao Q, Wang XY, Qiu SJ et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res 2009;15(3):971–979. doi: 10.1158/1078-0432.CCR-08-1608
[156] Cohen EE, Rosen LS, Vokes EE et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol 2008;26(29):4708–4713. doi: 10.1200/JCO.2007.15.9566
[157] Cohen Y, Xing M, Mambo E et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst 2003;95(8):625–627. doi: 10.1093/jnci/95.8.625
[158] Kebebew E, Weng J, Bauer J et al. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann Surg 2007;246(3):466–70; discussion 70–1. doi: 10.1097/SLA.0b013e318148563d
[159] van Veelen W, de Groot JW, Acton DS et al. Medullary thyroid carcinoma and biomarkers: past, present and future. J Intern Med 2009;266(1):126–140. doi: 10.1111/j.1365-2796.2009.02106.x
[160] Colombino M, Sperlongano P, Izzo Fet al. BRAF and PIK3CA genes are somatically mutated in hepatocellular carcinoma among patients from South Italy. Cell Death Dis 2012;3(1):e259. doi: 10.1038/cddis.2011.136
[161] Raymond E, Dahan L, Raoul JL et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011;364(6):501–513. doi: 10.1056/NEJMoa1003825
[162] Vijayvergia N & Cohen SJ. Personalized medicine in sporadic pancreatic cancer without homologous recombination-deficiency: are we any closer? J Gastrointest Oncol 2016;7(5):727–737. doi: 10.21037/jgo.2016.08.01
[163] Strippoli A, Rossi S, Martini M et al. ERCC1 expression affects outcome in metastatic pancreatic carcinoma treated with FOLFIRINOX: a single institution analysis. Oncotarget 2016;7(23):35159–35168. doi: 10.18632/oncotarget.9063
[164] Anderson A. Lynparza delays progression in metastatic pancreatic cancer patients with germline BRCA mutations. 2019. Available at: https://www.genomeweb.com/cancer/lynparza-delays-progression-metastatic-pancreatic-cancer-patients-germline-brca-mutations (accessed November 2019)
[165] Bellmunt J, Powles T & Vogelzang NJ. A review on the evolution of PD-1/PD-L1 immunotherapy for bladder cancer: the future is now. Cancer Treat Rev 2017;54:58–67. doi: 10.1016/j.ctrv.2017.01.007
[166] GenomeWeb. FDA approves Qiagen companion Dx for bladder cancer drug Balversa. 2019. Available at: https://www.genomeweb.com/regulatory-news/fda-approves-qiagen-companion-dx-bladder-cancer-drug-balversa (accessed November 2019)
[167] GenomeWeb. Genomic signatures offer insights into non-invasive bladder cancer. 2017. Available at: https://www.genomeweb.com/sequencing/genomic-signatures-offer-insights-non-invasive-bladder-cancer (accessed November 2019)
[168] Hurst CD, Alder O, Platt FM et al. Genomic subtypes of non-invasive bladder cancer with distinct metabolic profile and female gender bias in KDM6A mutation frequency. Cancer Cell 2017;32(5):701–715 e7. doi: 10.1016/j.ccell.2017.08.005
[169] McCusker M, Basset-Seguin N, Dummer R et al. Metastatic basal cell carcinoma: prognosis dependent on anatomic site and spread of disease. Eur J Cancer 2014;50(4):774–783. doi: 10.1016/j.ejca.2013.12.013
[170] Cirrone F & Harris CS. Vismodegib and the hedgehog pathway: a new treatment for basal cell carcinoma. Clin Ther 2012;34(10):2039–2050. doi: 10.1016/j.clinthera.2012.08.011
[171] Hanna A & Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer 2016;15(1):24. doi: 10.1186/s12943-016-0509-3
[172] Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol 2014;46(1):3–12. doi: 10.4103/0253-7613.124884
[173] Miettinen M & Lasota J. Gastrointestinal stromal tumours: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 2006;130:1466–1478. PMID: 17090188
[174] Rammohan A, Sathyanesan J, Rajendran K et al. A gist of gastrointestinal stromal tumors: a review. World J Gastrointest Oncol 2013;5(6):102–112. doi: 10.4251/wjgo.v5.i6.102
[175] Lennartsson J & Ronnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev 2012;92(4):1619–1649. doi: 10.1152/physrev.00046.2011
[176] National Institute for Health and Care Excellence. Imatinib for the adjuvant treatment of gastrointestinal stromal tumours. NICE guideline [TA326]. 2014. Available at: https://www.nice.org.uk/guidance/ta326 (accessed November 2019)
[177] National Institute for Health and Care Excellence. Sunitinib for the adjuvant treatment of gastrointestinal stromal tumours. NICE guideline [TA179]. 2009. Available at: https://www.nice.org.uk/guidance/ta179 (accessed November 2019)
[178] National Institute for Health and Care Excellence. Regorafenib for treating advanced gastrointestinal stromal tumours. NICE GID-TA10089. 2017. Available at: https://www.nice.org.uk/guidance/indevelopment/gid-ta10089 (accessed November 2019)
[179] Royal College of Obstetricians and Gynaecologists. Progress in cervical screening in the UK. Scientific Impact. 2016;1(paper no. 7):9. Available at: https://www.rcog.org.uk/globalassets/documents/guidelines/scientific-impact-papers/sip_7-progress-in-cervical-screening.pdf (accessed November 2019)
[180] Cancer Research UK. Soft tissue sarcoma incidence statistics. Available at: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/soft-tissue-sarcoma/incidence (accessed November 2019)
[181] Eds Fletcher CDM, Unni KK & Mertens F. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: World Health Organization: 2002
[182] Choe J & Riedel R. Making progress in a rare disease: emerging therapeutics in soft tissue sarcomas. F1000Research 2018;7. doi: 10.12688/f1000research.15868.1
[183] Steppan DA, Pratilas CA & Loeb DM. Targeted therapy for soft tissue sarcomas in adolescents and young adults. Adolesc Health Med Ther 2017;8(8):41–55. doi: 10.2147/AHMT.S70377
[184] Mertens F, Antonescu CR & Mitelman F. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer 2016;55(4):291–310. doi: 10.1002/gcc.22335
[185] Nakano K & Takahashi S. Translocation-related sarcomas. Int J Mol Sci 2018;19(12):3784. doi: 10.3390/ijms19123784
[186] Pollack SM, Ingham M, Spraker MB & Schwartz GK. Emerging targeted and immune-based therapies in sarcoma. J Clin Oncol 2018;36(2):125–135. doi: 10.1200/JCO.2017.75.1610
[187] Flaherty KT, Le DT & Lemery S. Tissue-agnostic drug development. 2017. Available at: https://meetinglibrary.asco.org/record/138072/edbook#fulltext (accessed November 2019)
[188] US Food and Drug Administration. FDA approves first cancer treatment for any solid tumor with a specific genetic feature. 2017. Available at: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm560167.htm (accessed November 2019)
[189] GenomeWeb. Melanoma BRAF mutation type may affect response to targeted treatments, immunotherapy. 2019. Available at: https://www.genomeweb.com/cancer/melanoma-braf-mutation-type-may-affect-response-targeted-treatments-immunotherapy (accessed November 2019)
[190] Merck. Merck’s KEYTRUDA® (pembrolizumab) plus pemetrexed (ALIMTA®) and platinum chemotherapy reduced the risk of death by half compared with chemotherapy alone as first-line treatment for advanced nonsquamous NSCLC in Phase 3 KEYNOTE-189 study. 2018. Available at: https://investors.merck.com/news/press-release-details/2018/Mercks-KEYTRUDA-pembrolizumab-Plus-Pemetrexed-ALIMTA-and-Platinum-Chemotherapy-Reduced-the-Risk-of-Death-by-Half-Compared-with-Chemotherapy-Alone-as-First-Line-Treatment-for-Advanced-Nonsquamous-NSCLC-in-Phase-3-KEYNOTE-189-Study/default.aspx (accessed November 2019)
[191] Le DT, Uram JN, Wang H et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596
[192] Yan L & Zhang W. Precision medicine becomes reality — tumor type-agnostic therapy. Cancer Commun (Lond) 2018;38(1):6. doi: 10.1186/s40880-018-0274-3
[193] Le DT, Durham JN, Smith KN et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;28;357(6349):409–413. doi: 10.1126/science.aan6733
[194] GenomeWeb. Breast cancer mutation signatures suggest mismatch repair subset. 2017. Available at: https://www.genomeweb.com/genetic-research/breast-cancer-mutation-signatures-suggest-mismatch-repair-subset (accessed November 2019)
[195] GenomeWeb. MMR deficiency ties to checkpoint blockade response explored in prospective study. 2017. Available at: https://www.genomeweb.com/sequencing/mmr-deficiency-ties-checkpoint-blockade-response-explored-prospective-study (accessed November 2019)
[196] Garber K. Tissue-agnostic cancer drug pipeline grows, despite doubts. Nat Rev Drug Discov 2018;17(4):227–229. doi: 10.1038/nrd.2018.6
[197] Ray T. Loxo oncology to seek FDA approval for pan-cancer drug in patients with TRK fusions. Chicago: GenomeWeb. 2017. Available at: https://www.genomeweb.com/sequencing/loxo-oncology-seek-fda-approval-pan-cancer-drug-patients-trk-fusions (accessed November 2019)
[198] Landman Y, Ilouze M, Wein Set al. Rapid response to larotrectinib (LOXO-101) in an adult chemotherapy-naive patients with advanced triple-negative secretory breast cancer expressing ETV6-NTRK3 fusion. Clin Breast Cancer 2018;18(3):e267–e70. doi: 10.1016/j.clbc.2017.11.017
[199] Ray T. Loxo’s pan-cancer drug garners FDA approval for patients with NTRK fusions. New York: GenomeWeb; 2018. Available at: https://www.genomeweb.com/regulatory-news/loxos-pan-cancer-drug-garners-fda-approval-patients-ntrk-fusions (accessed November 2019)
[200] Drilon A, Nagasubramanian R, Blake JF et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov 2017;7(9):963–972. doi: 10.1158/2159-8290.CD-17-0507
[201] Ray T. Industry interest in pan-cancer indications growing with FDA support despite challenges. 2019. Available at: https://www.genomeweb.com/cancer/industry-interest-pan-cancer-indications-growing-fda-support-despite-challenges (accessed November 2019)
[202] Houston D. Supportive therapies for cancer chemotherapy patients and the role of the oncology nurse. Cancer Nurs 1997;20(6):409–413. doi: 10.1097/00002820-199712000-00004
[203] Scotté F. The importance of supportive care in optimizing treatment outcomes of patients with advanced prostate cancer. Oncologist 2012;17(Suppl 1):23–30. doi: 10.1634/theoncologist.2012-S1-23
[204] Rewari B, Gupta S & Agarwal A. Supportive care in oncology. Journal, Indian Academy of Clinical Medicine 2004;5(1):38–46
[205] Lin CP, Hsu CH, Fu WM et al. Key opioid prescription concerns in cancer patients: a nationwide study. Acta Anaesthesiol Taiwan 2016;54(2):51–56. doi: 10.1016/j.aat.2016.05.002
[206] Martini G. What impact will personalised medicine have on pharmacy? Pharm J 2016;297(7891):58. doi: 10.1211/PJ.2016.20201328


