

Alamy
In 2004, Stephen Waxman, a neuroscientist at the Yale school of medicine in New Haven, Connecticut, was contacted by families, doctors and scientists from around the world who described cases of inherited pain. The condition, called inherited erythromelalgia (EM), otherwise known as man-on-fire syndrome, is incredibly rare. “Putting on a sweater, putting on shoes, going into a warm room, even mild exercise — they describe as feeling like hot lava being poured into their bodies,” says Waxman. His team had just published a paper that could explain why[1]
.
Meanwhile, Geoff Woods, a geneticist at University of Cambridge in the UK, was conducting his own investigations to try to explain why a family in Pakistan seemed to feel no pain at all. Woods had been told about a boy in the family who put knives through his arms as part of a street performance. In 2006, he published his findings on congenital inability to experience pain[2]
. A handful of similar families have since been identified, all of whom are otherwise normal, apart from lacking a sense of smell.
These seemingly opposite conditions are, in fact, caused by mutations of the same gene. The SCN9A gene encodes a voltage-gated sodium channel called Nav1.7, which is involved in propagating electrical signals along nerves. Waxman found that in patients with EM, the Nav1.7 channel is hypersensitive to stimulus, while Woods found it is non-functional in those who cannot feel pain.
Nav1.7 has implications not just for patients with EM, but for up to one in five adults who suffer with various types of chronic pain and rely on drugs that are either not effective enough, such as ibuprofen, or come with considerable side effects, such as the addictive properties of opioids.
“There is lots of hope that blocking this channel could temporarily knock out pain,” says Steve McMahon, a physiologist specialising in pain at King’s College London and director of the London Pain Consortium. Novel pain medicines are in great demand. And the beauty of the Nav1.7 channel is that it seems to selectively affect pain sensing while leaving other senses, such as touch, intact. There have now been several positive phase II clinical trials of Nav1.7 blockers and researchers are feeling optimistic. “You could say it’s a bit of a golden age for pain research,” says McMahon.
However, despite the genetic findings, there is debate about exactly how the channel works. One theory is that effectively blocking Nav1.7 increases levels of endogenous opioids and that adding a Nav1.7 blocker to opioid analgesia could dramatically cut the dose of opioid needed.
Drug development
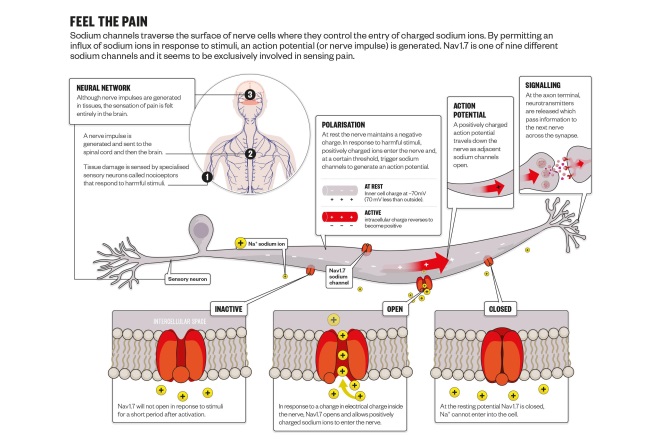
Voltage-gated sodium channels propagate signals along the nerve. When nerves are stimulated, there is a change in electrical potential and this triggers sodium channels in the membrane of nerve cells to open and allow positively charged sodium ions to enter the cell, changing the electrical charge from negative to positive and initiating an action potential that travels along the length of the nerve.

Stephen Waxman, a neuroscientist at the Yale school of medicine, is investigating why some families experience inherited pain
Since sodium channels were first identified in 1952, it has been implicit that they play a role in pain sensing because they play a role in all senses. Novocaine (procaine), for example, is a sodium channel blocker. “We all know we can get injected by the dentist and put the nerve to sleep but if you orally ingest Novocaine and introduce it into the brain you get central side effects, like double vision and sleepiness,” says Waxman. This is because Novocaine affects sodium channels in the central nervous system and the brain.
One of the most eagerly pursued questions in pain research had been whether there are certain sodium channels that play a role in the peripheral pain sensing neurons but do not play a significant role in the central nervous system, says Waxman. When he was studying to be a doctor in the 1970s, students were taught about “the” sodium channel. Now, there are nine known sodium channels, all with slightly different structures and functions.
And three of these channels provide the answer the researchers were looking for: Nav1.7, Nav1.8 and Nav1.9. They are all potential pain targets but Nav1.7 has attracted the most interest because of the dramatic genetic findings.
Since first being contacted by families with EM in 2004, Waxman’s research programme at Yale has become a worldwide hub for studying the genetics of inherited pain in humans. His laboratory has characterised more than a dozen Nav1.7 mutations. And in 2012 and 2014, he published research identifying the involvement of Nav1.8[3]
and Nav1.9[4]
in certain painful neuropathic conditions.
Around half a dozen large pharmaceutical companies and a handful of smaller ones are working to develop drugs that target the Nav1.7 channel. And at least three candidate drugs have entered clinical trials, including one from Pfizer and one from Convergence, both based in the UK, and another from British Columbia-based Xenon, in collaboration with California-based Genentech. Waxman has been working closely with both Pfizer and Convergence, although he adds that he has no personal stake in either of the drugs.
Leading the race to bring a drug to market is Convergence with its sodium channel blocker raxatrigine, which it is developing for a type of chronic pain called trigeminal neuralgia, a condition that causes attacks of electric shock-like facial pain that can be felt regularly for months at a time.
In November 2015, Convergence reported internally the results of a phase II trial with 29 patients, which found that pain intensity scores were 50% lower for patients receiving raxatrigine than for those receiving placebo (P=0.0009). In 2016, the company plans to conduct a phase III 12-week double-blind placebo controlled trial. It is also testing the molecule for its effectiveness against sciatica in phase II trials, and in the future could apply it to other forms of neuropathic pain.

Simon Tate is chief scientific officer at Convergence, which is developing sodium channel blocker raxatrigine for a type of chronic pain called trigeminal neuralgia
Convergence began developing raxatrigine prior to the genetic discoveries in 2004 and 2006. “We had a head start because we had already been working on this for a number of years,” says Simon Tate, chief scientific officer at Convergence. The work began when Tate was at GlaxoSmithKline before Convergence was formed as a spin-off company in 2010. Convergence has since been acquired by Biogen, based in Cambridge, Massachusetts. Tate says that when the genetic findings “broke” the molecule was tailored to Nav1.7, but it is not completely selective for this channel over other sodium channels.
However, raxatrigine has selectivity in another way. It targets the most active sodium channels and therefore the areas with the most pain signalling, something that Tate calls “block-on-demand”. Sodium channels cycle through three different states: open, closed and inactive. Raxatrigine stabilises it in the inactive state. The theory is that the more sodium channels are held inactive, the harder it is to propagate the pain signal, says Tate.
But because the molecule is not entirely specific for the Nav1.7 channel, it also interacts with sodium channels in the central nervous system. Despite this, Tate says it has a well-tolerated safety profile.
Although promising, the potential of Nav1.7 as a drug target has not yet been proven clinically. Data for the Pfizer drug, also designed to block Nav1.7, were presented at the International Association for the Study of Pain in Buenos Aires in October 2014. EM is a good testbed for the effectiveness of Nav1.7 blockers because the effect can be attributed directly to that channel. In five patients with EM, Pfizer’s drug seemed to lessen heat-induced pain more than placebo, but full details are yet to be released. Xenon and Genentech have two Nav1.7 blockers in phase I trials and say both “GDC-0276 and GDC-0310 are promising candidates”.
Source: Alisdair Macdonald
Nav1.7 is one of nine different sodium channels that traverse the surface of nerve cells where they control the entry of charged ions. By permittingan influx of positive ions in response to stimuli, an action potential (or nerve impulse) is generated that acts as a pain signal
Four theories
The story of Nav1.7 may be more complex than it initially seemed. The pharmacogenetic data have not been replicated by clinical findings, says McMahon. The problem, he says, is that no one fully understands the mechanism of Nav1.7.
“It’s a beautiful target in theory. We know it’s critical because of the mutations but we’re not sure why,” says McMahon. One of the crucial questions that needs to be answered is how much the Nav1.7 channels have to be blocked to have an effect. “Is it 50%, 90%? It’s a perplexing mystery at the moment,” he says.
There are four theories about how Nav1.7 works and none are mutually exclusive. The first, which is well established, is that it is simply involved in propagating the pain signal along the nerve. However, even in patients without a functioning Nav1.7 channel, pain signals are still generated, they just do not seem to be interpreted as pain.
The second theory is that Nav1.7 channels regulate the sensitivity of the neuron to stimuli. In chronic pain, neurons become more sensitive to stimuli and evidence suggests that Nav1.7 plays a significant role. The third theory is that Nav1.7 channels are involved in the release of a neurotransmitter at the junction between nerves. Neurotransmitters carry information from one nerve to the next. The idea is that when Nav1.7 does not function, the pain element of the signal is cut short at the first synapse. There is some evidence for this, which needs to be replicated and validated, says Waxman.
Ion channels are known to have an influence on gene expression. So the fourth theory is that Nav1.7 alters the expression of other genes involved in pain perception. This was proposed by John Wood at UCL, London, who recently published the findings of a study that showed Nav1.7 influences the expression of endogenous opioid peptides[5]
. When Nav1.7 function is lost, the level of opioid peptides goes up.
The opioid peptides are part of an innate system in the body that produces analgesic and euphoric effects, for example, endorphins released during exercise. The opioid peptides implicated in the mechanism of Nav1.7 are called enkephalins, which regulate pain pathways in the spinal cord. An increase in enkephalins could therefore be contributing to the loss of pain experienced in patients without a functional Nav1.7 channel.
Perhaps enkephalins are the missing part of the puzzle. A potent and specific Nav1.7 blocker, called Protoxin II, failed to produce the same analgesic effects in rats as having a non-functional Nav1.7 channel[6]
. But by combining a Nav1.7 blocker with a very low dose of opioids, Wood says he has produced dramatic pain relief from diabetic neuropathy and rheumatoid arthritis in mice[5]
. “If you could 100% block the channel, then perhaps [eventually] the opioids would go up on their own,” he says. He suggests that Nav1.7 may need to be blocked more efficiently and for a long period of time to induce changes in opioid levels, which could explain why clinical findings do not quite match the genetic evidence. Even more compelling evidence to substantiate this theory is that using naloxone to block the action of endogenous opioids in a person with a Nav1.7 loss-of-function mutation seemed to allow them to feel pain for the first time[5]
.
The real joy of this story, says Wood, is that this strategy could allow doses of opioid analgesics to be dramatically cut if combined with a Nav1.7 blocker. This could make the side effects of opioids, such as addiction, less troublesome. “It looks like endogenous opioids are in play for decades without any harmful effects, so this could provide a strategy for really cutting down massively on the opioid doses,” he explains. All of this is yet to be shown in humans, but the animal model studies suggest that the combination could make a huge difference.
Next, Wood’s team want to find out exactly how this opioid system is regulated and whether it is central or peripheral in the nervous system. He plans to begin the first proof-of-concept studies in humans in 2016, using one of the Nav1.7 blockers that has been approved for clinical trials in combination with one of the many available opioids.
Waxman, however, says the jury is still out on this theory because many ion channels alter gene expression. The question is, how unique or important is Nav1.7 in this regard? “It’s a provocative idea, I don’t know if it’s correct but if it is, it’s important,” he says. McMahon is also sceptical of this theory.
Other strategies
Regardless of which theory is correct, Nav1.7 is still a highly prized target and it is not the only one being pursued to tackle chronic pain. McMahon identified the involvement of another pathway in pain signalling in 1995, he found that an increase in nerve growth factor protein (Ngf)[7]
, released by immune cells during the process of inflammation, can contribute to the development of a chronic pain state in nearby nerves.
There are high levels of Ngf in patients with chronic inflammatory pain. As expected, it seems to be a particularly good target for painful inflammatory conditions, such as osteoarthritis. Around three or four companies are developing drugs against Ngf, including a collaboration between Pfizer and Lilly that has produced a monoclonal antibody called tenazumab. “Blocking Ngf works in the clinic, but it has potential side effects,” says McMahon. In 2010, a phase III trial of tenazumab was suspended by the US Food and Drug Administration (FDA) when it seemed that it might be linked with osteonecrosis (bone death), resulting in joint replacement. However, after a review by the FDA, phase III trials were allowed to continue in March 2015.
Nav1.7 and Ngf are perhaps the most advanced strategies in development, but there are others. In chronic pain, the neural networks in the brain can be altered, becoming more receptive to negative stimuli. So researchers in Northwestern University in Chicago, Illinois, are trying to prevent this remodelling of brain circuitry by using dopamine antagonists that stimulate the reward system in the brain, with promising results in mice[8]
. “All of a sudden, like buses, there seems to be lots of good news at once,” says McMahon, who says the dopamine study sounds “plausible”.
As yet, it is hard to say which strategy is the most promising. Waxman, who has dedicated his career to researching the role of sodium channels in pain, says he couldn’t possibly comment. But adds coyly: “I guess I’m voting with my feet.”
References
[1] Cummins TR, Dib-Hajj SD & Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. The Journal of Neuroscience 2004;24:8232–8236. doi: 10.1523/jneurosci.2695-04.2004
[2] Cox JJ, Reimann F & Nicholas AK. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006;444:894–898. doi: 10.1038/nature05413
[3] Faber CG, Lauria G, Merkies IS et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA 2012;109(47):19444–19449. doi: 10.1073/pnas.1216080109
[4] Huang J, Han C, Estacion M et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 2014;137:1627–1642. doi: 10.1093/brain/awu079
[5] Minett M, Pereira V, Sikandar S et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nature Communications 2015;6:8967. doi: 10.1038/ncomms9967
[6] Schmalhofer WA, Calhoun J, Burrows R et al. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Molecular Pharmacology 2008;74:1476–1484. doi: 10.1124/mol.108.047670
[7] McMahon S, Bennet D, Priestley J et al. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nature Medicine 1995;1:774–780. doi: 10.1038/nm0895-774
[8] Ren W, Centeno MV, Berger S et al. The indirect pathway of the nucleus accumbens shell amplifies neuropathic pain. Nature Neuroscience 2016;19:220–222. doi: 10.1038/nn.4199
You may also be interested in
