Steve Gschmeissner / Science Photo Library
Within the next two years, scientists are likely to begin clinical trials of a drug aimed at alleviating debilitating side effects from irinotecan, a chemotherapy agent used in advanced colon and pancreatic cancer. This drug doesn’t target biochemical processes in human cells, nor is it an antimicrobial that kills ‘foreign’ organisms. Instead, for the first time, it works by modulating the activity of the microbiome, the 100 trillion bacteria that inhabit the human gut.
“Our goal is to keep patients on the drugs that are keeping them alive. And we’re doing that through the microbiome, which is a new paradigm,” says Matthew Redinbo, a biochemist at the University of North Carolina in Chapel Hill. Redinbo led studies demonstrating in mice that manipulating the microbiome in this way can prevent the severe diarrhoea that often limits the use of irinotecan and related molecules[1]
. He’s now creating a spin-off company, Symberix, to commercialise the drug.
The work would represent the first clinical application of insights into how the symbiotic bacteria that help maintain our health interact with the pharmaceuticals we take to improve our health. It’s a field of study so new that no one even agrees on what to call it. Candidates include pharmacomicrobiomics and pharmacometabonomics, among others.
And yet, in principle it isn’t new at all. “The general concept that the microbiome is involved in drug metabolism is actually fairly old,” says Peter Turnbaugh, a microbiologist at the University of California, San Francisco. “This idea has been around for decades.”
But over the past few years, the idea has come into the spotlight. Several studies have shown the wide variety of effects the microbiome can have on drug metabolism. Microbes can activate or inactivate drugs, generate toxic byproducts of drug metabolism, and alter drug metabolism by human cells in both direct and indirect ways. In many ways, the field is a natural extension of pharmacogenomics, the study of how variations in human genes contribute to individual differences in drug metabolism and effectiveness. In addition, the Human Microbiome Project —established to characterise the human microbiota and analyse their role in human health and disease — has focused attention on the big variations between the normal microbiota of different individuals.
You can give two people the same drug and they will respond differently. And it’s not because of their genes but because of the genes of the microbes they carry
“You can give two people the same drug and they will respond differently. And it’s not because of their genes but because of the genes of the microbes they carry,” says Ramy Aziz, a microbiologist in the Faculty of Pharmacy at Cairo University. As a result, Aziz and other scientists say, the development of truly personalised medicine will have to consider humans as a superorganism, composed of ten times as many bacterial cells as human ones, and will have to take into account the idiosyncrasies not just of a single human genome but of the one to two thousand bacterial genomes that come along with it.
Fortunately — and perhaps the biggest reason for the current flowering of this area of research — the advent of high-throughput sequencing, advanced bioinformatics, and techniques such as metagenomics are enabling scientists to get a handle on this complexity.
Historical mysteries
It is bioinformatics that has enabled researchers to confirm hunches and test hypotheses about microbes and drugs that in some cases are decades old. Around 30 years ago, scientists traced differences in how individuals process the cardiac medicine digoxin to a gut bacterium called Egerthella lenta, which is capable of inactivating, or reducing, digoxin[2]
. Only people who harbour E. lenta in their gut are able to inactivate the drug, scientists found. Yet not everyone with E. lenta does so.
“And so there was this real mystery,” says Turnbaugh. It was a mystery with enormous therapeutic consequences, because those who inactivate the drug may get little therapeutic benefit from it, while a too-high dose given to someone who doesn’t could prove toxic.
Turnbaugh and his colleagues solved the mystery by sequencing RNA from E. lenta. They identified a pair of genes, called the cgr operon, that work together to metabolise digoxin[3]
.
“If you compare different E. lenta strains, you find that only some of them carry the operon,” says Turnbaugh. The group also showed that E. lenta carriers who have a high proportion of bacteria with the cgr operon metabolise a high proportion of digoxin, while those colonised primarily with strains that lack the operon inactivate very little of the drug.
Now, Turnbaugh and his colleagues are investigating how to manipulate this process — for example, by blocking E. lenta’s action to ensure that more active digoxin reaches the heart. The cgr operon is less active in the presence of the amino acid arginine, and the researchers have found that mice colonised with E. lenta fed a high-protein diet have higher blood digoxin levels compared with those given a protein-free diet. Next, they plan to see whether arginine supplementation alone can achieve the same effects.
Redinbo’s work on preventing side effects from irinotecan also rests on relatively familiar observations about the microbiome. Scientists have long known that the liver processes many drugs by adding a sugar called glucuronic acid. This both detoxifies the molecule and ‘tags’ it for transport to the intestine and elimination.
Meanwhile, around half of the microbes that inhabit the human gut produce an enzyme called beta-glucuronidase, which enables them to scavenge that glucuronic acid from these molecules and reactivate them, a process called enterohepatic recycling. In the case of a highly toxic drug like irinotecan, such recycling could be disastrous. “This hypothesis had been out in the literature for a while — that it was the bacterial beta-glucuronidases that were reactivating irinotecan and causing the toxicity,” says Redinbo.
So Redinbo and his colleagues set out to test this hypothesis by looking for a drug that would inhibit bacterial beta-glucuronidase, without harming the microbes themselves or interfering with the action of the body’s own versions of the enzyme. The researchers screened thousands of chemicals and found several that could prevent irinotecan-induced diarrhoea in mice, they reported in 2010[1]
.
Redinbo collaborated with Urs Boelsterli, a toxicologist at the University of Connecticut in Storrs, on further mouse studies to show that inhibitors of bacterial beta-glucuronidase also prevent ulceration of the colon by nonsteroidal anti-inflammatory drugs (NSAIDS), a class of widely used painkillers[4]
. “Once we did it a second time with NSAIDs, it feels quite real to me,” Redinbo says. “We can control the microbiome without killing it.”
Once we did it a second time with NSAIDs, it feels quite real to me — we can control the microbiome without killing it
Since the body processes many drugs by this pathway, beta-glucuronidase inhibitors could have wide applications for alleviating drug side effects.
Indirect effects
The microbiome can have subtler effects on drug dynamics, too. “It’s not just about the gut microbial metabolism, there are all sorts of indirect ways that microbes can manipulate host metabolism,” says Turnbaugh.
In 2009, researchers led by Jeremy Nicholson, chair of biological chemistry at Imperial College, traced differences in processing of another common pain reliever, paracetamol, to the microbiome. They showed that people with a relatively high proportion of a chemical called p-cresol in their urine metabolise paracetamol differently than people with low levels of urinary p-cresol.[5]
P-cresol is produced by a bacterium called Clostridium difficile. To the human body, p-cresol looks a lot like paracetamol, and the liver processes it the same way — by adding a sulfur-containing group to the molecule. Too much p-cresol keeps the enzymes involved in this process busy, bumping metabolism of paracetamol on to other biochemical pathways and producing the differences that Nicholson and his colleagues observed.
The results suggest that people with high levels of p-cresol produced in their gut might experience liver toxicity from lower doses of paracetamol compared with people whose gut microbiota yield only low levels of p-cresol.
More broadly, the results may help to explain why drug side effects are not the same across all individuals, says Ian Wilson, chair of drug metabolism and molecular toxicology at Imperial College London, who collaborates frequently with Nicholson but was not involved in the paracetamol work. Pharmacologists often struggle to explain so-called ‘idiosyncratic toxicity,’ the serious, 1-in-10,000 adverse reactions that lead to black box warnings on drugs. “One clear cause of idiosyncracy, potentially, is differences in the gut microflora,” says Wilson.
One clear cause of idiosyncracy, potentially, is differences in the gut microflora
The microbiome may also influence the effectiveness of a drug by shaping the activity of other body systems, especially the immune system. Two studies published in 2013 suggest that gut microbiota play a role in the effectiveness of cancer chemotherapy by modulating inflammatory and immune responses that target tumours.
In one study, a team of French scientists showed that cyclophosphamide, an anticancer drug that acts by coordinating the body’s immune system to fight a tumour, is less effective in mice that lack gut microbes or have had their microbiome knocked down by large doses of antibiotics[6]
.
The researchers also conducted experiments to show the mechanism behind these effects: cyclophosphamide alters the composition of the gut microbiota and increases permeability of the intestinal wall. In response, certain species of bacteria move from the gut into nearby lymph nodes, where they guide the maturation of immune cells called T cells that go on to attack the tumour.
In the second study, led by scientists at the US National Cancer Institute (NCI), researchers showed that antibiotic-treated mice or those without gut microbiota don’t respond as well to a type of cancer treatment called CpG-oligonucleotide immunotherapy or to the platinum-based drugs oxaliplatin and cisplatin, compared with mice that have healthy gut microbiota[7]
.
Both studies were conducted in mice that had cancer cells injected under their skin, and it’s not yet clear what the results mean for humans undergoing cancer treatment. Still, the results hint that it might be possible to increase the effectiveness of chemotherapy by manipulating the microbiome. For example, in the future, oncologists might think twice about giving their patients preventative antibiotics before a course of chemotherapy. Or they may even administer helpful microbes — the presence of bacterial groups called Alistipes and Ruminococcus was correlated with chemotherapy effectiveness in the NCI-led study.
Microbiome-host interaction
The gut microbiota consists of approximately 100 trillion microbial cells. The metabolic activities of these microbes expand host metabolic capabilities by activating or inactivating drugs, generating toxic byproducts of drug metabolism, and altering drug metabolism by human cells in both direct and indirect ways.
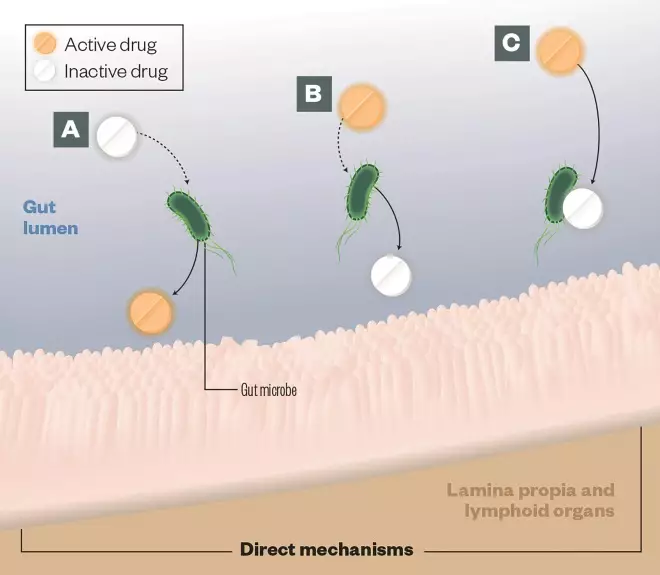
A) Activation: Gut microbes can convert prodrugs into active drugs, (eg, sulfasalazine)
B) Deactivation: Gut microbes can encode enzymes that detoxify drugs, eg, digoxin is inactivated by the gut-residing Actinobacterium Eggerthella lenta
C) Direct binding: Gut microbes can bind directly with drugs, compromising their bioavailability, eg, levadopa is converted to dopamine in the gut, preventing it from reaching the brain
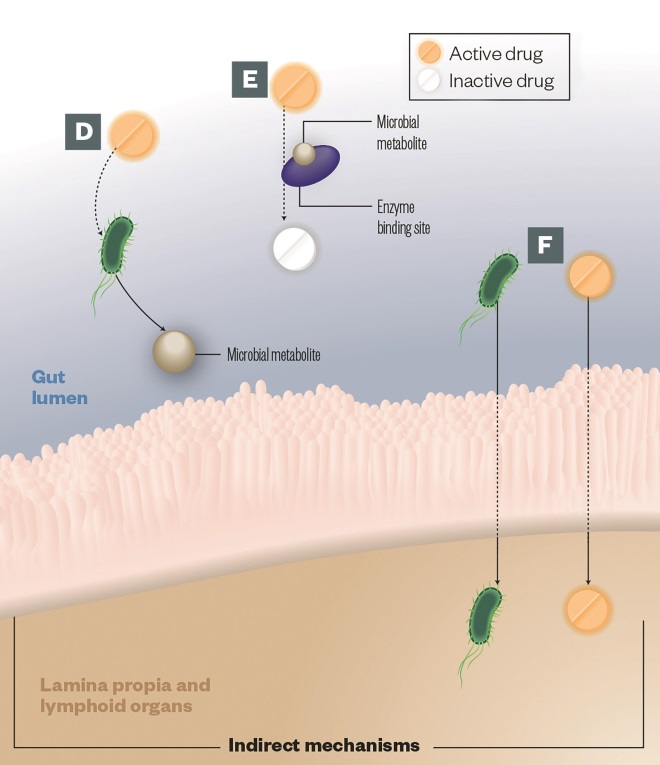
D) Intermediates: Some metabolic pathways share steps between host enzymes and those encoded by gut microbes. Intermediate microbial products can lead to pathogenesis; the best known example of this is not a therapeutic drug but a toxin called melamine
E) Altered kinetics: Gut microbial metabolites compete with drugs for enzyme-binding sites, leading to alterations in the drug efficacy or toxicity, e.g. paracetamol
F) Immune response: The gut microbes can impact drug efficacy by stimulating the host immune system, e.g. cyclophosphamide increases gut permeability leading microbes to translocate into the lymph nodes
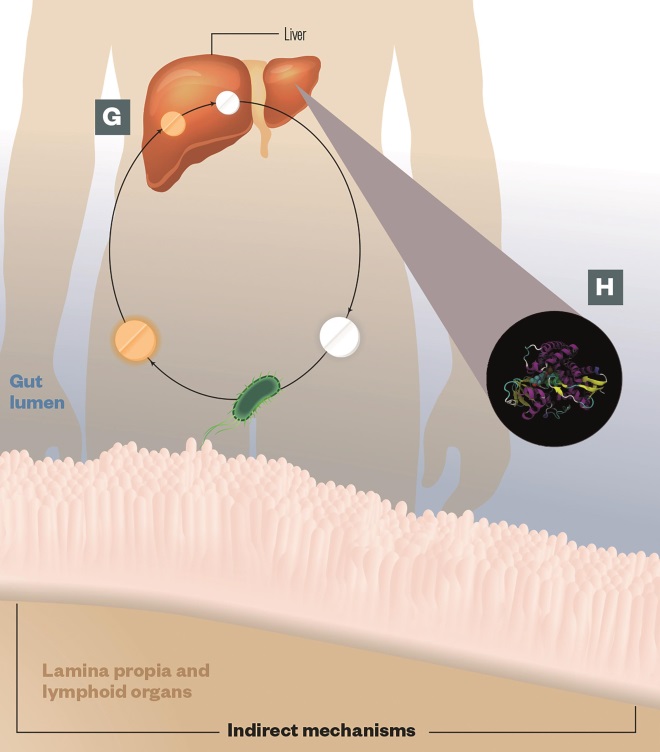
G) Enterohepatic cycling: The liver processes many drugs by adding glucuronic acid, which detoxifies the drug and ‘tags’ it for transport to the intestine and elimination. Gut microbes produce an enzyme called beta-glucuronidase, which enables them to scavenge the glucuronic acid and reactivate the drug, leading to toxicity, e.g. irinotecan
H) Altered gene expression: Gut microbes can alter the hepatic expression of key host enzymes involved in drug metabolism, e.g. CYP450 (pictured), leading to alterations in drug efficacy or toxicity
Source: Journal of Clinical Investigation 2014
Systematic knowledge
Approximately 60 drugs are known to be affected by gut microbes, according to the PharmacoMicrobiomics database, an online, open-source resource that represents the first systematic collection of emerging knowledge in the field[8]
.
The real number is probably much higher, scientists agree. But even for those drugs where some microbial effect is known to occur, details are often sketchy. “In most cases, the literature will just say that if you take this drug it will be modified in one way or another by the intestinal bacteria,” says Aziz, who runs the database. “In most cases we don’t know which bacterium.”
This still represents an enormous increase in knowledge since 2007, when Aziz started working on the database with one of his classes at Cairo University in Giza, Egypt. At the time, “there were a few scattered studies”, some of them decades old, says Aziz.
Eight years later, Aziz confidently predicts that microbiome analysis will become a routine part of deciding on a treatment regimen for patients, the same way genetic analysis has become common in planning cancer treatment.
If that’s to happen, the field — whatever it ends up being called — will have to welcome many more researchers. “There is still little being done in this area relative to other topics that are linked to the microbiome,” says Turnbaugh. “Hopefully that will change and we’re at the beginning of an upswing.”
References
[1] Wallace BD, Wang H, Lane KT et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 2010;330:831–835. doi:10.1126/science.1191175.
[2] Saha JR, Butler VP, Neu HC et al. Digoxin-inactivating bacteria: identification in human gut microbiota. Science 1983;220:325–327.
[3] Haiser HJ, Gootenberg DB, Chatman K et al. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta . Science 2013;341:295–298.
[4] LoGuiduce A, Wallace BD, Bendel L et al. Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. Journal of Pharmacology and Experimental Therapeutics 2012;341:447–454. doi:10.1124/jpet.111.191122.
[5] Clayton TA, Baker D, Lindon JC et al. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proceedings of the National Academy of Sciences USA 2009;106:14728–14733. doi:10.1073/pnas.0904489106.
[6] Viaud S, Saccheri F, Mignot G et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013;342:971–976.
[7] Lida N, Dzutsev A, Stewart CA et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013;342:967–970.
[8] Rizkallah MR, Gamal-Eldin S, Saad R et al. The PharmacoMicrobiomics Portal: A database for drug-microbiome interactions. Current Pharmacogenomics and Personalized Medicine 2012;10:195–203.