Wikimedia Commons
In this article you will learn:
- How to interpret an ECG to detect long QT syndrome
- The possible causes of long QT syndrome
- How to manage patients’ medicines to reduce risk of long QT syndrome
Heart contractions are caused by a series of electrical impulses that originate from the sino-atrial (SA) node, located in the right atrium. These impulses travel through the atria, which causes atrial contraction and stimulates the atrioventricular (AV) node. The electrical impulses are then sent from the AV node via the bundle of His, the left and right bundle branches and the purkinje fibres to stimulate ventricular contraction.
This electrical activity can be monitored using an electrocardiogram (ECG) to identify any problems in transmission. One of the most important potential problems is long QT syndrome.
The QT interval is the time between the start of the Q wave and end of the T wave (shown in figure 1). The ECG spike known as the QRS complex senses ventricular contraction, and the T wave represents ventricular relaxation, with the QT interval being a measure of the total time from contraction to relaxation of the ventricles.
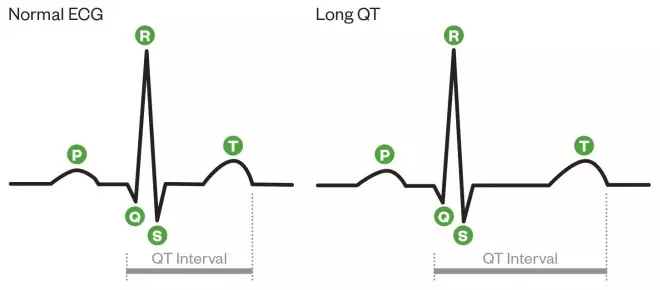
Normal ECG versus prolonged QT
Normal ECG versus prolonged QT
Long QT syndrome can be caused by a number of factors, including electrolyte imbalances and genetic mutations in ion channels. It can also be caused by medicines, and patients taking new drugs are monitored for long QT syndrome during clinical trials and following licensing.
Over the past 20 years, a number of non-antiarrhythmic drugs (for example, cisapride in 2000 and terfenadine by 2005) have been withdrawn from the UK market, or have had their product characteristics revised (for example, citalopram in 2011 and ondansetron in 2012) due to post-marketing reports of prolonged QT interval. In September 2014, domperidone was reclassified in the UK as POM because of concerns over QT prolongation.
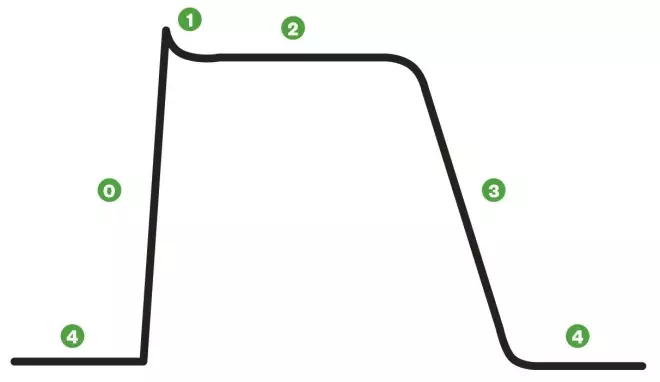
A cardiac action potential
Non-pacemaker action potentials, such as those of the atrial and ventricular cells, are sometimes called fast-response action potentials. These action potentials have a true resting potential, a fast depolarisation phase and a prolonged plateau, followed by repolarisation.
Phase 0: Cells are depolarised to a threshold voltage by an action potential in an adjacent cell, and the transient increase in intracellular sodium via fast sodium channels causes cellular depolarisation.
Phase 1: is the initial repolarisation, caused by the opening of potassium channels. At the same time, L-type calcium channels allow inward movement of calcium, prolonging the action potential and causing the plateau effect (phase 2). This does not occur in action potentials found in skeletal muscle.
Phase 3: As calcium channels deactivate, repolarisation occurs as potassium channels open and potassium diffuses out of the cell. If depolarisation is triggered during phase 3, this can lead to ventricular tachycardia or fibrillation.
Primary symptoms of long QT syndrome may include palpitations, syncope and seizures. However, many patients do not have symptoms and the syndrome is only diagnosed after a cardiac event, or if a patient is prompted to have an ECG after a family member has died from sudden cardiac death.
Long QT syndrome is a known risk factor for ventricular arrhythmias, in particular torsades de pointes. Torsades de pointes can cause syncope, seizures or sudden cardiac death due to insufficient cardiac output.
The QT interval is measured using a 12-lead ECG. However, the interval varies depending on a patient’s heart rate, and measurements must be corrected to take this change into account. This is expressed as corrected QT interval (QTc).
There are several formulae used for QTc, but the most common (known as Bazett’s formula) is to divide the distance between the start of the QRS complex and the end of the T wave by the square root of the preceding RR interval (see ‘Calculating QTc’).
Calculating QTc
1) Calculate the preceding RR interval, measured in seconds. This is equivalent to 60 divided by heart rate (in beats per minute)[1]
.
2) Measure the distance between the start of the QRS complex and the end of the T wave (QT interval).
3) Divide the QT interval by the square root of the RR interval.
QTc is equivalent to the QT interval at a heart rate of 60 beats per minute (RR interval = 1 second). If the heart rate is 100 beats per minute (RR interval = 60/100 = 0.6s), a measured QT interval of 360ms would produce a QTc of around 465ms.
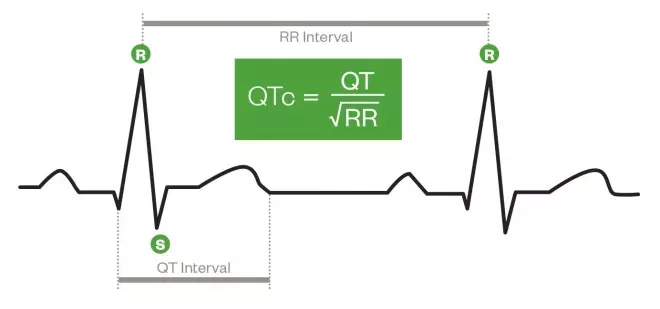
Calculating QTc – different measurements of an ECG trace
A normal QTc is typically less than 440ms in men and less then 460ms in women. The QTc interval in men decreases at puberty and then gradually increases as androgen levels fall
; this is also the reason women typically have a longer QT interval than men.
There is no formal definition of what is considered a prolonged QTc interval. As a guide, a ‘borderline’ QTc interval is between 440ms and 500ms. A QTc of more than 500ms is clinically significant and indicates that the patient is at increased risk of arrhythmia.
Diagnosis of long QT syndrome
Diagnosis of long QT syndrome is based on the results of an ECG and the patient’s clinical and family history. Many people with long QT syndrome do not have symptoms, and the condition may be diagnosed following an incidental finding on an ECG. At least three measurements of QTc should be made on lead II of a 12-lead ECG to confirm the diagnosis.
Genetic testing of first-degree family members is routinely offered for all patients with suspected long QT syndrome. There are at least 13 gene mutations known to cause a long QT interval, the majority of which are dominant alleles.
Other diagnostic tests include checking electrolytes (potassium and magnesium) and thyroid function tests to eliminate secondary reasons for repolarisation abnormalities. If diagnosis is not confirmed initially, patients may undergo cardiac stress testing to measure QTc at elevated heart rates[2]
.
Risk factors for long QT syndrome include:
- female sex
- increasing age
- liver or renal impairment
- family history of congenital long QT syndrome
- bradycardia
- pre-existing cardiovascular disease
- electrolyte imbalance
- concurrent administration of interacting drugs[3]
Causes of long QT syndrome
Acquired long QT syndrome is most commonly caused by electrolyte imbalances (hypokalaemia and hypomagnesaemia) and medicines (see ‘Medicines known to prolong QT interval’). Myocardial infarction, raised intracranial pressure or cranial trauma, bradycardia, hypothyroidism and severe hypothermia are also possible causes.
Medicines can affect QT interval by prolonging ventricular repolarisation, inhibiting the metabolism of drugs that prolong the QT interval, or by causing hypokalaemia and hypomagnesaemia (for example, diuretics).
Medicines known to prolong QT interval
| Class | Common examples |
|---|---|
Antiarrhythmic | Class I: quinidine, procainamide, disopyramide, flecainide Class III: sotalol, amiodarone (rare), dronedarone |
Antimicrobial | Erythromycin, clarithromycin, trimethoprim, ketoconazole, itraconazole, chloroquine |
Antihistamine | Terfenadine (no longer available), diphenhydramine |
Other drugs | Amitriptyline, cisapride (no longer available), chlorpromazine, domperidone, glibenclamide, ondansetron, haloperidol, citalopram, lithium |
A comprehensive list can be found at www.qtdrugs.org
Marked QT prolongation, often accompanied by torsades de pointes, occurs in 1-10% of patients receiving antiarrhythmic drugs that can cause acquired long QT syndrome, and more rarely in non-cardiovascular drugs that can prolong the QT interval[2]
.
Conditions that inhibit the metabolism or excretion of medicines that prolong the QT interval (for example, liver or renal impairment) can also increase a patient’s risk of acquired long QT syndrome. Prolonged QT intervals have been found in patients with diabetic ketoacidosis, and are associated with the level of ketosis, even in the absence of electrolyte abnormalities. Patients with anorexia nervosa are also at increased risk of prolonged QT intervals.
Congenital long QT syndrome is caused by genetic mutations affecting sodium or potassium ion channels, which can lead to prolonged ventricular repolarisation. This can cause ventricular arrhythmias and syncope. Congenital long QT syndrome is thought to affect 1 in 7,000 people in the UK population[4]
. The exact prevalence is unknown, as many people remain asymptomatic and undiagnosed.
The mean age for symptoms to first occur is 12 years, and patients usually present with syncope triggered by a sudden increase in sympathetic activity such as intense emotions, rigorous exercise or sudden noises (for example, alarm clocks and telephones).
The three most common forms of congenital long QT syndrome are long QT 1 (LQT1), long QT 2 (LQT2), and long QT 3 (LQT3), which account for around 75% of all known cases[5]
. The risk of experiencing cardiac events increases if QTc is longer than 500ms in all forms of congenital long QT syndrome.
LQT1 is caused by a genetic mutation in the KCNQ1 gene, which encodes a potassium channel responsible for termination of the plateau phase of the action potential. Symptoms are most commonly caused by exercise, and usually first occur in adolescence and early adulthood[5]
.
LQT2 is the most prevalent subtype of congenital long QT syndrome, and is caused by genetic mutations in the KCNH2 gene. This gene encodes another potassium channel that terminates the action potential (which is also the channel most frequently blocked by medicines). Possible triggers include intense emotions and loud noise[5]
.
LQT3 is caused by genetic mutations in the SCN5A gene, which encodes a subunit of the sodium channel causing sodium influx and depolarisation. Prolonged QT events are more common during rest or sleep. Patients with an LQT3 mutation have a lower risk of cardiac symptoms such as syncope and dizziness than those with an LQT1 or LQT2 mutation, but are at higher risk of sudden death[5]
.
Mutations in SCN5A can also cause Brugada syndrome, cardiac conduction disease, and dilated cardiomyopathy.
The lifetime risk of a fatal arrhythmia in those with congenital long QT syndrome is around 13%. Patients at high risk of fatal arrhythmia include: men and women with LQT1 and a QTc of more than 500ms; all men with LQT2 and women with LQT2 and a QTc of more than 500 ms; all men and women with LQT3[6]
.
Management of long QT syndrome
Torsades de pointes (a severe manifestation of prolonged QT interval) is a medical emergency. It usually terminates spontaneously, although it may result in ventricular fibrillation. Patients with prolonged torsades de pointes are treated with direct-current cardioversion. Patients with recurrent bursts of torsades de pointes, or torsades de pointes following a cardiac arrest, are treated with magnesium 2g intravenous infusion followed by temporary cardiac pacing[5]
.
Acquired long QT syndrome should be suspected in patients presenting with torsades de pointes or ventricular fibrillation. In such patients, medicines known to prolong QTc must be stopped immediately and emergency treatment started.
Underlying causes should also be identified and stopped in patients with a QTc of more than 500ms. Magnesium should be given without waiting for blood results, and patients should be given potassium if blood levels are less than 4mmol/l. Thereafter, potassium levels should be maintained at 4.5-5mmol/l. Long-term ion replacement is not usually necessary[6]
.
Patients with known acquired long QT syndrome should have a baseline ECG when medicines that can prolong the QT interval are prescribed. This should be repeated when the medicine reaches a steady state, and then as appropriate every three to six months. There is a 5% increased risk of arrhythmic events for every 10ms increase in QTc[7]
.
All patients at high risk of acquired long QT syndrome (such as those taking medicines known to cause prolonged QT, or those who have previously been diagnosed with long QT syndrome) should be monitored with an ECG when starting a medicine that can cause a prolonged QT interval.
The lowest effective dose should always be prescribed for any medicine that can prolong the QT interval, as the risk of QT prolongation is always greater if higher doses are used. For example, citalopram 20mg can cause a mean change in QTc of 7.5ms, whereas citalopram 60mg can increase QTc by 16.7ms. For this reason, the maximum daily dose of citalopram is restricted to 40mg for adults and 20mg for patients aged over 65 years[8]
.
ECGs should be repeated when clinically indicated (for example, when increasing the dose, switching to or adding a medicine that can prolong the QT interval, when ECG or electrolyte abnormalities are detected, or when symptoms suggesting arrhythmia occur). Patients should be told to watch for symptoms of arrhythmias (for example, palpitation, light headedness and syncope) during treatment[9]
.
If a patient develops symptoms, pharmacists should assess the severity of the symptoms and consider the medicine’s indication, then use their clinical judgement as to whether the patient should stop the medicine or seek further advice.
Congenital long QT syndrome is managed by preventing the sudden increases in sympathetic activity that may trigger arrhythmias, which can reduce the risk of cardiac events. All patients should be advised to avoid known precipitating factors, particularly exercise that significantly raises heart rate.
All patients should be treated with beta-blockers, although they are more useful in LQT1 than LQT3[5]
. This includes patients who are asymptomatic, including children, to reduce the risk of sudden cardiac death. Despite optimising the dose of beta-blockers, 20-25% of patients will remain symptomatic and at high risk of sudden death. These patients may be treated with left cervical-thoracic sympathetic neural denervation, a surgical procedure that blocks sympathetic input to the heart.
Patients at high risk of sudden cardiac death (QTc of more than 550ms), or those whose first event is a cardiac arrest requiring resuscitation, may have an implantable cardioverter defibrillator inserted by the surgical team. These devices do not prevent life-threatening ventricular arrhythmias, but will shock the heart to terminate the arrhythmia. Recurrent shocks can interfere with a patient’s quality of life, and careful patient section and counselling is important before the operation.
Patients with congenital long QT syndrome should avoid medicines known to prolong the QT interval or that can deplete potassium or magnesium. Patients should be told to inform healthcare professionals that they have long QT syndrome.
Community pharmacists should be aware of over-the-counter medicines which are contraindicated in long QT syndrome. These include over-the-counter cough and cold remedies containing the antihistamine diphenhydramine and the antifungal fluconazole. Patients can take potassium or sodium supplements if they have been told to increase their dietary intake.
Patients with congenital long QT syndrome should be referred to their GP if they have an acute illness involving diarrhoea or vomiting, as these symptoms can deplete electrolyte levels.
Clare Thomson MPharm, MRPharmS, DipGenPP is a rotational cardiac pharmacist and
Paul Wright MPharm, MRPharmS, MSc is lead cardiac Pharmacist at University College London Hospitals NHS Foundation Trust.
References
[1] Anon. American Heart Association Recommendations for the Standardization and Interpretation of the Electrocardiogram. Circulation. 2009,119:241–250.
[2] European Society of Cardiology Clinical Practice Guidelines. Guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death [Online]. 2006 Available at: escardio.org (accessed 10 August 2014).
[3] Ramrakha P & Hill J. Oxford Handbook of Cardiology. 2nd Edition. Oxford: University Press 2012.
[4] Arrhythmia Alliance, Heart Rhythm Charity. Long QT syndrome [Online]. Available at: heartrhythmcharity.org.uk (accessed 10 August 2014)
[5] Ackerman M & Johnson J. QTc: how long is too long? Br J Sports Med. 2009;43:657–662.
[6] Priori SG, Schwartz PJ, Napolitano C et al. Risk stratification in the long-QT syndrome N Engl J Med 2003;348:1866–1874.
[7] Taylor D, Paton C, Kapur S. The South London and Maudsley NHS Foundation Trust. Prescribing Guidelines in Psychiatry. 11th Edition. Chichester: Wiley Blackwell 2012.
[8] Medicines Healthcare and Regulatory Agency Drug Safety Update. Citalopram and escitalopram: QT interval prolongation — new maximum daily dose restrictions. Volume 5, Issue 5 (online). 2011. Available at: mhra.gov.uk (accessed 24 August 2014).
[9] Abdelmawla N & Mitchell A. Sudden cardiac death and antipsychotics, Part 2. Advances in Psychiatric Treatment (online) 12 p. 100–109. 2006. Available at: http://apt.rcpsych.org (accessed 24 August 2014).
You might also be interested in…

The impact of pharmacist-led medication intervention on patient perception and satisfaction following new acute coronary syndrome diagnosis prior to hospital discharge
NHS increases funding for inclisiran although uptake remains ‘slower than expected’