KATERYNA KON/SCIENCE PHOTO LIBRARY
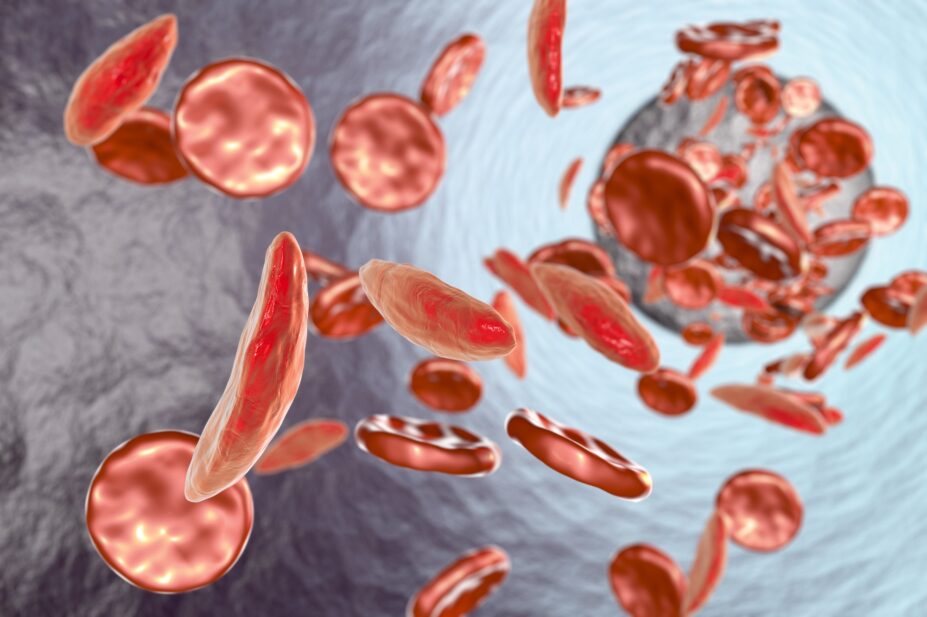
Sickle cell disease (SCD) is a general term for a group of inherited red blood cell disorders. Normal red blood cells have a biconcave disc shape that is flexible and can easily pass through blood vessels. However, those with SCD have abnormalities in their haemoglobin owing to genetic mutations, resulting in their red blood cells becoming ‘sickle’ shaped rather than round[1]. This inhibits the cells’ ability to carry oxygen and can therefore restrict the amount of oxygen received by the limbs and organs. The cells are also rigid and sticky, causing the red blood cells to stick together, reducing their ability to move through the blood vessels easily. This can lead to blockages, which cause the patient significant pain[2].
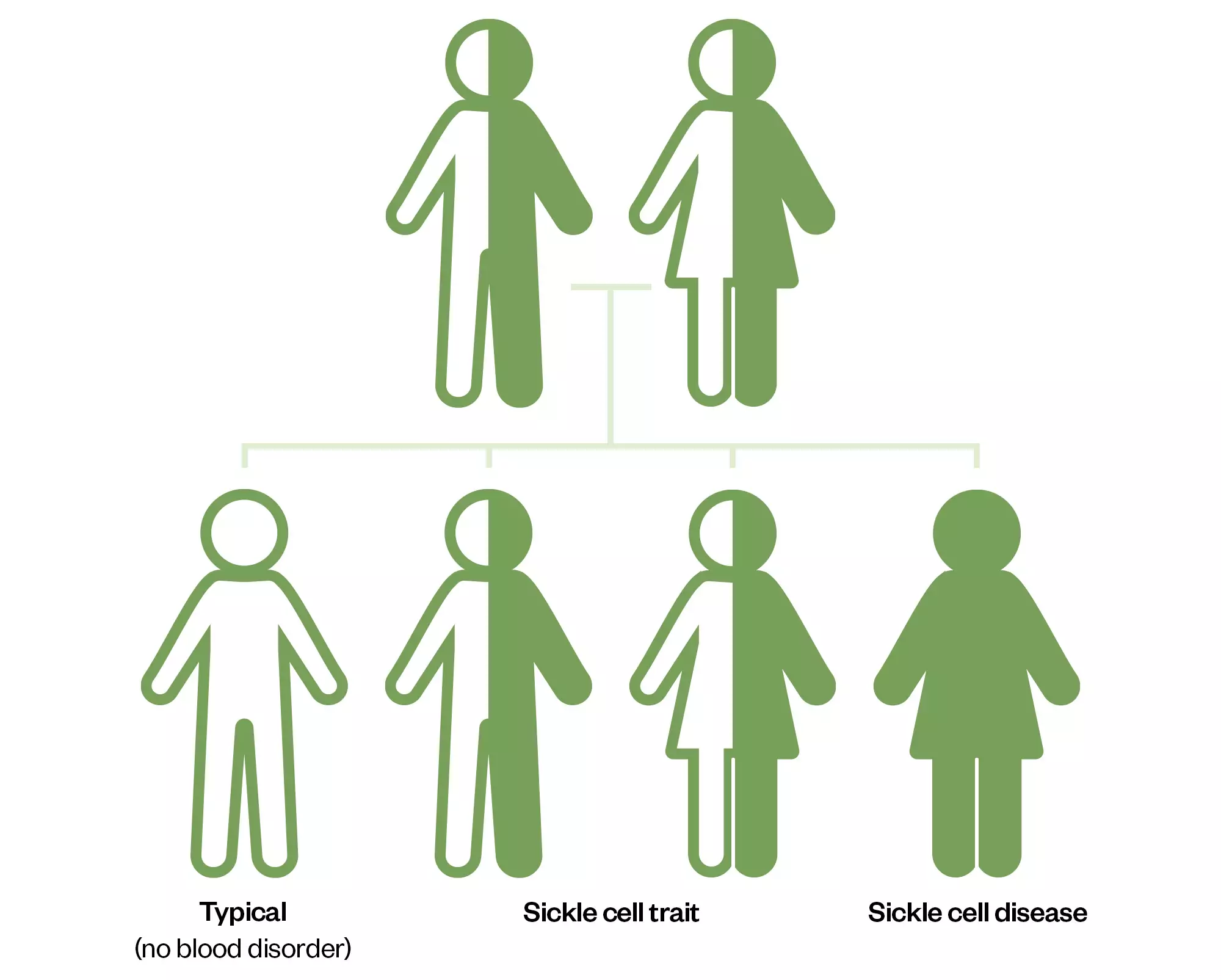
The most common types of SCD are HbSS, HbSC and HbS beta thalassaemia. HbSS SCD occurs when an individual inherits two genes that code for haemoglobin ‘S’, the abnormal form of haemoglobin that causes the sickle shape of the red blood cell. This type is often referred to as sickle cell anaemia and is the most severe form. HbSC SCD occurs when an individual inherits one haemoglobin ‘S’ and one haemoglobin ‘C’ gene. Both cause abnormal red cell shapes, but this is usually a milder form of SCD. HbS beta thalassaemia occurs when an individual inherits one ‘S’ gene and one beta thalassaemia gene; both genes cause abnormal red blood cell shapes[3].
The Pharmaceutical Journal
There are thought to be around 14,000 people with SCD in the UK, although accurate and up-to-date information is lacking, meaning there could be considerably more[4]. In England, 1 in every 2,000 live births are affected by SCD. It is most common in people of black African and black Caribbean heritage, but also affects people from the Middle East, Mediterranean and South America[1].
Pathway to diagnosis
In the UK, SCD is included in the national screening programme for newborn babies. On day five after birth, a newborn blood spot test is used to screen for different congenital disorders. The SCD screening programme screens for genetic carriers of SCD, thalassaemia, sickle cell trait and other haemoglobin disorders[5]. If the blood tests are not clear, genetic testing can determine what type of SCD the patient has. All pregnant women are screened for sickle cell trait; if they find out they are a carrier, the other biological parent can be tested too.
In addition, prenatal screening should be carried out in babies born to parents from an ethnic group where there is high prevalence of SCD. Prenatal screening can help expecting parents make more informed choices during the pregnancy and help them prepare if there is a potential the baby will need treatment for SCD. Amniocentesis can be considered as a diagnostic test for genetic conditions, during which a sample of amniotic fluid or tissue from the placenta is taken[6]. It carries risks but it is more accurate than prenatal screening blood tests to find out which type of sickle cell disease the baby might have. According to the NHS, the risk of miscarriage from amniocentesis is estimated to be 1 in 100 (1%) after 15 weeks of pregnancy; the risk is increased before 15 weeks[7].
Presentation
SCD can have diverse clinical presentations, ranging from acute generalised pain to early-onset strokes, leg ulcers, and premature mortality resulting from multi-organ failure. A defining feature of SCD is intense pain that is unpredictable and episodic. The intensity of the pain and frequent hospitalisations largely account for the pervasive negative impact on patients’ health-related quality of life[8].
Risks and complications
SCD is associated with both acute and long-term complications that necessitate a comprehensive, multidisciplinary approach.
People with SCD face an elevated risk of developing complications in their lungs, heart and kidney function, primarily owing to reduced blood and oxygen supply to these vital organs. Manifestations of organ damage can encompass a range of symptoms, including but not limited to breathlessness, irregular heart rhythms, nausea, swelling of extremities and jaundice[9].
Pain is the most frequent acute complication and substantially affects health-related quality of life. Pain episodes are often triggered by infection, exposure to cold and dehydration. Owing to the vaso-occlusive effects of sickled red blood cells, there is a risk of further conditions such as deep vein thrombosis, renal or hepatic crises and acute chest syndrome[10].
Acute vaso-occlusive episodes, colloquially known as sickle cell crises or pain crises, are the leading cause for seeking immediate medical attention[10]. During an episode, sickle cells obstruct smaller blood vessels, impeding normal blood flow. This causes severe pain, which can manifest in various body parts; most commonly, in the hands, feet, chest and back[11]. The duration of these painful episodes varies, and an episode can emerge suddenly and unexpectedly. Known triggers include dehydration, extreme temperature fluctuations, consumption of alcohol, smoking and use of illicit drugs; however, crises may also occur without any apparent provocation.
SCD is also associated with hypersplenism, a condition in which the sequestration of sickle red blood cells in the spleen leads to splenomegaly. The reduction in splenic function renders patients more susceptible to life-threatening infections, such as influenza, meningitis and pneumonia[12]. To mitigate this risk, immunisations and regular low-dose prophylactic antibiotics are advised. Vaccines against pathogens, such as haemophilus influenzae type B (HiB), pneumococcal and meningococcal vaccinations, are essential to provide adequate protection.
Patients who experience left upper quadrant pain, rapid breathing and an elevated heart rate should promptly seek medical attention, as these are symptoms of splenic sequestration. Any rise in body temperature or feeling unwell should also be promptly discussed with a healthcare provider.
Acute treatment options
According to the National Institute for Health and Care Excellence (NICE), a clinical assessment should be conducted with SCD patients who present with signs and symptoms of a sickle cell crisis[13]. All patients who present to A&E or an acute care setting with an acute painful episode should be offered analgesia within 30 minutes of presentation[13].
Patients who attend A&E with moderate pain, who have not had any medication at home, can initially be given a non-steroidal anti-inflammatory drug (NSAID), such as ibuprofen or naproxen, and weak opioids, such as codeine or low-dose oral morphine, if not contraindicated[13,14]. Patients with severe pain, or moderate pain that continues after previous analgesia, should be offered strong opioids up front, such as morphine or oxycodone. This can be administered orally or via subcutaneous injection, depending on patient choice and the clinical situation[14].
Effectiveness of pain relief should be assessed every 30 minutes from initial administration of medication, until satisfactory pain relief has been achieved[14]. There is no way to objectively measure pain, so the assessment is usually based on pain scale ratings. However, ratings are subjective and lower pain ratings can often be overlooked in terms of providing pain relief[15]. If pain continues after repeated doses of strong analgesia, then patient-controlled analgesia should be considered. Supportive treatment includes regular paracetamol and NSAIDs (unless contraindicated) plus opioids if needed. As opioids can inhibit gastric emptying and peristalsis, which results in opioid-induced constipation, patients are given laxatives. In addition, because of the wide distribution of receptors that opioids influence, the patient should be monitored for nausea and vomiting and offered an antiemetic to reduce this. IV fluids may be given to patients who are not able to tolerate oral fluids, or as part of their plan[16]. IV fluids are given to patients during a pain crisis to slow the sickling process, which can reduce the duration and amount of pain for the patient; however, care should be taken to not fluid overload the patient, as fluid could build up in the heart or lungs, causing further complications[16].
Long-term treatment options
There are limited long-term treatment options for patients with SCD. Hydroxycarbamide (hydroxyurea) is an oral medication that has been shown to reduce the frequency and severity of acute sickle cell crisis and acute chest syndrome (a complication of SCD)[17]. The exact mechanism of action of the medicine is unknown; however, in SCD, it is thought to elevate foetal haemoglobin, which interferes with the polymerisation of sickle haemoglobin, preventing the sickling of red blood cells. Hydroxycarbamide ameliorates several symptoms of SCD; however, side effects include skin pigmentation, thinning hair, low white cell and platelet count and low sperm count[17].
Blood transfusion or automated red cell exchange are other ways of reducing the severity of acute and chronic complications of SCD. It is generally offered to patients who are at risk of serious complications, such as stroke, and patients whose condition is not controlled by hydroxycarbamide[17]. After one year of blood transfusions, an iron-chelating agent such as Exjade should be considered to reduce the risk of iron overload[18].
Crizanlizumab is an IV treatment that can be administered alone or alongside hydroxycarbamide[19]. Its mechanism of action is binding to P-selectin on endothelial cells and platelets, which prevents them interacting with P-selectin glycoprotein ligand 1 on platelets and red blood cells. This results in blood components being less likely to ‘stick’ together, reducing the likelihood of vaso-occlusive crisis. This monoclonal antibody is only recommended to prevent recurrent vaso-occlusive crises in people aged 16 years and over and if conditions in the managed access agreement are followed. Currently, an application for this treatment must be made by a specialised haemoglobinopathy team. It is administered every four weeks, except for the first two treatments, which are two weeks apart. Crizanlizumab was the first new treatment in 20 years for SCD; however, as of November 2023, the MHRA has approved a new gene therapy, Casgevy (exagamglogene autotemcel; Vertex Pharmaceuticals), which has the potential to cure SCD by editing the faulty genes that cause the red cells to be sickle shaped[20,21].
Stem cell and bone marrow transplants are currently the only established cure for SCD, but they are not performed often because of the significant risks involved; these include infection, graft-versus-host disease and organ injury from the conditioning regimen, which involves immunosuppressive medications and low-dose radiotherapy[22]. Some of these complications can be fatal. The overall success rate of stem cell and bone marrow transplants has been estimated to be ~93%[23].
The patient experience: Frankie’s story
It can be difficult to imagine the experiences a patient with SCD may have to go through. The following story is that of a fictional patient, Frankie, but is based on the lived experiences of a patient with SCD. Take the time now to use what you have learned above, plus your own experiences, to work through the clinical case below.
Patient overview
Often, Frankie starts the day feeling OK, but then becomes unwell and ends up in A&E with a sickle cell crisis. Frankie has a successful career and strives to live as normally as possible.
After a persistent pain episode lasting a few days, Frankie presented to A&E. The following hospital notes were recorded.
Reflective questions
- Review the treatment plan recorded in the hospital notes. What counselling points do you think should be discussed with Frankie?
- Based on the information contained in the notes, what management considerations can you identify?
- How can the pharmacist potentially provide support for Frankie during their presentation to A&E?
Frankie’s perspective: a visual story

Xin Yi Teo
Sickle cell and thalassaemia community services are an excellent source of support, contact, advice, and advocacy. Services could be delivered by an individual or comprise a whole team, including nurses (matrons), counsellors and healthcare professionals. Frankie has a good connection with their community matron and will ring them and let them know that they have had to go to hospital.
Although the receptionists are very understanding, and get things done quickly, Frankie feels that they will have to wait a long time to be called in to be reviewed.

Xin Yi Teo
Frankie is reviewed in A&E and a plan has been made for treatment of their pain crisis.

Xin Yi Teo

Xin Yi Teo
Frankie is discharged from hospital, although tired they are glad to be home and in their own bed and their pain is tolerable for now. Frankie will update their sickle cell community matron.
Reflective questions
- How did reading Frankie’s perspective affect your responses to the previous questions? Were you able to identify any further management considerations as a result?
- How could we ensure Frankie is offered the correct treatments in the desired timeframe(s)?
- How can we improve Frankie’s perception of pain management during the acute phase of treatment?
- How can we better measure Frankie’s pain during their pain crisis? For example, what does satisfactory pain mean to Frankie? What questions could be asked instead of using a numerical pain scale?
- What support or information could be provided to Frankie regarding long-term analgesia?
- What can you implement into your own practices to better support patients?
Best practice for pharmacists
Pharmacists are well placed to help patients with SCD in several ways.
Medication
Pharmacists can provide regular counselling to ensure adherence to the patients’ treatment regimens. Reinforcing counselling points to SCD patients may help them understand better how they can help manage their condition. Counselling points could include specific points about their medication, side effects or health and lifestyle.
Lifestyle
Some health and lifestyle counselling points pharmacists can advise on is to drink plenty of fluids daily to avoid dehydration, take care in extreme temperature conditions (try to avoid sudden temperature changes), try to avoid alcohol as this can lead to dehydration and try to avoid smoking as this can trigger acute chest syndrome.
Optimisation
When a pharmacist interacts with a SCD patient, they should ensure the medication is optimised for the individual patient, so they have appropriate doses of pain relief, the medication is effective and the benefits outweigh the risks. In addition, a pharmacist can help with adverse side effects from medications and how to manage them.
Holistic
It is important for pharmacists to provide holistic care, such as asking the patient about their mental wellbeing. This is especially important for people with chronic conditions, such as SCD, as they can experience struggles that have a significant impact on their quality of life. It is therefore essential pharmacists can signpost patients to other help and guidance where necessary.
Conclusion
SCD is a chronic condition with many complications that not only significantly affect the patient’s quality of life, but have the potential to be life-threatening.
For an acute vaso-occlusive crisis, NICE has issued clear guidance, which includes the need for first-dose analgesia to be administered within 30 minutes of presentation. Treatment should be person-centred and take into consideration the patient’s preferences on next steps.
Long-term treatment options are limited, but currently consist of hydroxycarbamide, blood transfusions, iron-chelating agents, the monoclonal antibody crizanlizumab and, most recently, the gene therapy Casgevy.
Illustrations by: Xin Yi Teo – BA (Hons) Illustration student, School of Art and Media, University of Brighton
- This article was amended on 3 September 2026 to clarify that hydroxycarbamide is a long-term treatment option
- 1What is Sickle Cell Disease (SCD)? British Society for Haematology. 2023. https://b-s-h.org.uk/about-us/news/what-is-sickle-cell-disease-scd (accessed January 2024)
- 2Webb J, Kwiatkowski JL. Stroke in patients with sickle cell disease. Expert Review of Hematology. 2013;6:301–16. https://doi.org/10.1586/ehm.13.25
- 3What is Sickle Cell Disease? Centers for Disease Control and Prevention. 2023. https://www.cdc.gov/ncbddd/sicklecell/facts.html (accessed January 2024)
- 4How common is sickle cell disease? National Institute for Health and Care Excellence. 2021. https://cks.nice.org.uk/topics/sickle-cell-disease/background-information/prevalence/ (accessed January 2024)
- 5Scenario: Screening. National Institute for Health and Care Excellence. 2021. https://cks.nice.org.uk/topics/sickle-cell-disease/management/screening/ (accessed January 2024)
- 6Significant haemoglobinopathies: A guideline for screening and diagnosis. British Society for Haematology. 2023. https://b-s-h.org.uk/guidelines/guidelines/significant-haemoglobinopathies-a-guideline-for-screening-and-diagnosis (accessed January 2024)
- 7Risks: Amniocentesis. NHS. 2022. https://www.nhs.uk/conditions/amniocentesis/risks/ (accessed January 2024)
- 8Inusa B, Hsu L, Kohli N, et al. Sickle Cell Disease—Genetics, Pathophysiology, Clinical Presentation and Treatment. IJNS. 2019;5:20. https://doi.org/10.3390/ijns5020020
- 9Bender M, Carlberg K. Sickle Cell Disease. National Library of Medicine. 2022. https://www.ncbi.nlm.nih.gov/books/NBK1377/ (accessed January 2024)
- 10Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4. https://doi.org/10.1038/nrdp.2018.10
- 11Serjeant GR, Ceulaer CDE, Lethbridge R, et al. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol. 1994;87:586–91. https://doi.org/10.1111/j.1365-2141.1994.tb08317.x
- 12Houwing ME, de Pagter PJ, van Beers EJ, et al. Sickle cell disease: Clinical presentation and management of a global health challenge. Blood Reviews. 2019;37:100580. https://doi.org/10.1016/j.blre.2019.05.004
- 13Sickle cell disease: managing acute painful episodes in hospital. National Institute for Health and Care Excellence. 2012. https://www.nice.org.uk/guidance/cg143 (accessed January 2024)
- 14Standards for the Clinical Care of Adults with Sickle Cell in the UK. Sickle Cell Society. 2018. https://www.sicklecellsociety.org/wp-content/uploads/2018/05/Standards-for-the-Clinical-Care-of-Adults-with-Sickle-Cell-in-the-UK-2018.pdf (accessed January 2024)
- 15Anie KA, Grocott H, White L, et al. Patient self-assessment of hospital pain, mood and health-related quality of life in adults with sickle cell disease. BMJ Open. 2012;2:e001274. https://doi.org/10.1136/bmjopen-2012-001274
- 16Okomo U, Meremikwu MM. Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. Cochrane Database of Systematic Reviews. 2017;2021. https://doi.org/10.1002/14651858.cd005406.pub5
- 17Howard J, Telfer P. Sickle Cell Disease in Clinical Practice. Springer 2015.
- 18Mobarra N, Shanaki M, Ehteram H, et al. A Review on Iron Chelators in Treatment of Iron Overload Syndromes. Int J Hematol Oncol Stem Cell Res. 2016;10:239–47.
- 19Crizanlizumab — A Simple Guide. Sickle Cell Society. 2023. https://www.sicklecellsociety.org/crizanlizumab/ (accessed January 2024)
- 20NICE recommends first treatment in two decades for sickle cell disease. National Institute for Health and Care Excellence. 2021. https://www.nice.org.uk/news/article/nice-recommends-first-treatment-in-two-decades-for-sickle-cell-disease#:~:text=For%20the%20first%20time%20in,people%20aged%2016%20or%20over (accessed January 2024)
- 21UK medicines regulator approves world-first gene-editing treatment for blood disorders. Imperial College Healthcare NHS Trust. 2023. https://www.imperial.nhs.uk/about-us/news/uk-medicines-regulator-approves-world-first-gene-editing-treatment-for-blood-disorders (accessed January 2024)
- 22Scenario: Prevention of complications. National Institute for Health and Care Excellence. 2021. https://cks.nice.org.uk/topics/sickle-cell-disease/management/prevention-of-complications/ (accessed January 2024)
- 23Bolaños-Meade J, Brodsky RA. Blood and marrow transplantation for sickle cell disease: overcoming barriers to success. Current Opinion in Oncology. 2009;21:158–61. https://doi.org/10.1097/cco.0b013e328324ba04