This content was published in 2010. We do not recommend that you take any clinical decisions based on this information without first ensuring you have checked the latest guidance.
The metabolism of drugs occurs through basic chemical reactions as soon as the administered compound comes into contact with enzymes that are capable of altering its chemical structure. Conversely, a drug’s stability after administration is due mainly to its lack of transformation by the body’s enzymes. However, many drugs are susceptible to some form of chemical decomposition, be it through the interaction with enzymes or through improper storage and use, and such degradation often leads to a loss of potency.
A working knowledge of the functional groups within drug molecules, those that are likely to be susceptible to metabolism and those more likely to be resistant should be useful when counselling patients on the storage and use of their prescribed medicines.
A common example is dispensing medicines into monitored dosage systems (MDS), where medicines are removed from manufacturers’ packaging and placed in an environment where stability is unknown. Similarly, it is useful to have knowledge of functional groups whenever alternative therapeutic agents within a given chemical class are being considered as part of a medicines use review.
Chemical reactions
Most drugs are small organic molecules. The only differences between one drug and another are the number and type of functional groups present, and the connectivity between these groups. This is true of all organic molecules and, as such, there is little difference between drugs and other small organic molecules, such as those found in food and household products. The exception is that drugs have been specifically designed and optimised (over many years and costing hundreds of millions of pounds) (PJ, 11 September 2010) to have the desired pharmacological profile to enable them to treat a disease. With this in mind, it should therefore not be surprising that drugs undergo chemical reactions in much the same way as other organic molecules under set conditions.
The main chemical reactions that affect the stability of a drug are oxidation and hydrolysis. Oxidation involves the removal of electrons from a molecule (or the addition of oxygen) and such reactions can be initiated by light, heat or certain trace metals. Despite oxidation being a relatively common pathway for drug decomposition, it has not been studied in as much detail as hydrolysis since oxidative degradation can often be reduced to acceptable levels by storing susceptible drugs in the absence of light (eg, use of amber vials) and oxygen (eg, store under nitrogen or argon), or by using antioxidants in the formulation. Hydrolysis is the more common pathway for drug breakdown and is the mechanism that we will concentrate on here with regard to both chemical stability and metabolism.
Hydrolysis
Hydrolysis means the reaction of a molecule with water resulting in the cleavage of a chemical bond within that molecule. Although there are a large number of functional groups that are susceptible to hydrolysis, esters and amides are the most common ones found in drugs prone to hydrolysis. Figure 1 outlines the mechanism that accounts for the reaction of an ester (X=OR) or an amide (X=NR1R2, where R, R1 and R2 can be any arbitrary structure) resulting in the cleaved reaction products of a carboxylic acid and either an alcohol (XH=ROH) or an amine (XH=R1R2NH), respectively.
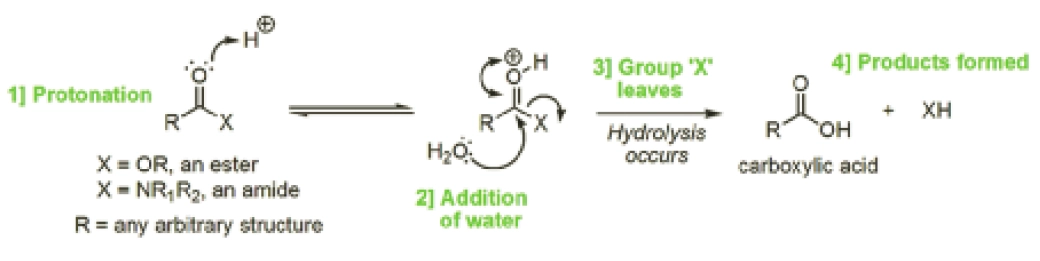
Oxygen can draw electrons towards itself more strongly than carbon, so the C=O double bond in the ester or amide becomes polarised, leaving the oxygen slightly negatively charged and the carbon slightly positively charged. As a result of this polarisation, the electrons on water’s oxygen atom are attracted to the slightly positive charge of the aforementioned carbon atom (unlike charges attract one another), resulting in hydrolysis (Figure 1, indicated by the bottom arrow at 2). This polarisation and subsequent interaction is enhanced if the carbonyl oxygen is protonated (as shown at 1 in Figure 1).
The reaction of water with esters proceeds more rapidly than with amides, and these reactions can be catalysed by both acid and base. The rate is dependent on the pH of the aqueous solution. Similarly, the in vivo metabolism of esters and amides in drugs via a hydrolysis mechanism is catalysed by hydrolytic enzymes present in various tissues and in plasma. The reason for the difference in the rates of ester and amide hydrolysis lies in the structural difference between these two functional groups. The ester contains an oxygen atom, whereas the amide possesses a nitrogen atom at the same position. This difference results in the carbon atom of the carbonyl group (C=O) being even more positively charged in the ester than in the amide and, as such, there is a greater attraction between that carbon and a water molecule. The opposite is true for the amide because the presence of the nitrogen atom in amides lessens the positive charge on the carbonyl carbon atom and thus it is less able to attract incoming water molecules.
This difference in the hydrolysis of esters and amides is exemplified with lidocaine and procaine (Figure 2).
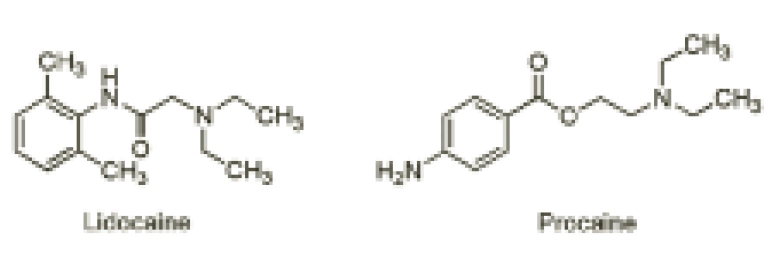
The ester-containing local anaesthetic procaine is now seldom used. Although it is a good local anaesthetic, its effect is short-lasting because its ester group is rapidly hydrolysed. Conversely, lidocaine contains an amide bond, which is less readily hydrolysed than procaine’s ester. As such, it is more stable to the effects of hydrolysis and this, in addition to lidocaine’s more bulky structure, makes it a longer-acting local anaesthetic.
Furthermore, the attention-deficit hyperactivity disorder drug methylphenidate (Figure 3) contains a methyl ester that is readily hydrolysed by hydrolytic enzymes to yield ritalinic acid as the major (inactive) urinary metabolite in humans.
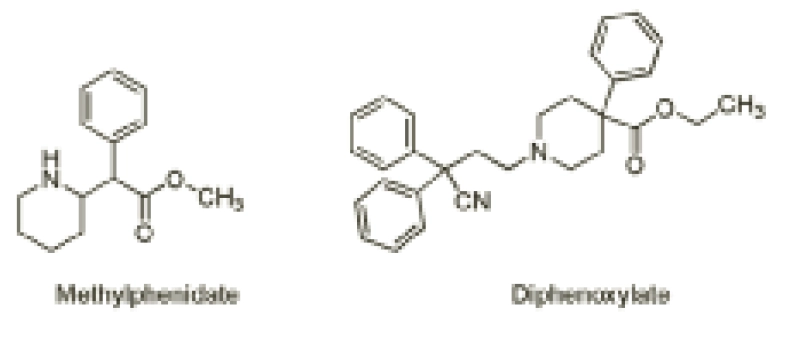
Such a readily hydrolysable functional group poses a problem since it renders the drug inactive and, therefore, the drug needs to be administered more than once daily to maintain therapeutic levels. As such, a great deal of research into modified-release formulations has resulted in once-daily Concerta XL and Equasym XL as a means to address the non-compliance associated with self-administration during school hours.
On the other hand, hydrolysis can also be helpful. The antimotility drug co-phenotrope contains the ester diphenoxylate (Figure 3), which is readily hydrolysed in humans to the related carboxylic acid, diphenoxylic acid, a five-times more potent antidiarrhoeal agent than the parent ester.
Prodrug strategy
Salicylic acid is a good painkiller. However, the presence of the bare alcohol moiety causes gastric bleeding. Such an effect is overcome by masking this alcohol group as an ester — giving us aspirin — which is later hydrolysed in the body to free the active drug. Such an approach is known as a prodrug strategy. The hydrolysis of aspirin also produces acetic acid as a product. In the poor storage of aspirin tablets, a smell of vinegar that pervades over time is a sign that hydrolysis is occurring.
Another prodrug example includes the angiotensin-converting enzyme inhibitor enalapril, whose ester is hydrolysed in vivo to generate the active carboxylic acid derivative enalaprilate (enalaprilate being used as an intravenous form of enalapril for hypertensive emergencies).
Type I statins, such as simvastatin, which contain a cyclic ester (a lactone), and are hydrolysed by body enzymes to produce the ring-opened, pharmacologically active hydroxy-acid common to these cholesterollowering drugs.
Amide-containing drugs
As previously mentioned, amide-containing drugs are also susceptible to hydrolysis, but it occurs at a much slower rate than the hydrolysis of esters. An example is the hydrolysis of the strained cyclic-amide ring (amides that are part of a ring structure are known as lactams) of b-lactam antibiotics. It is routine to prepare aqueous paediatric suspensions of penicillin antibiotics immediately before dispensing them since they are insufficiently stable to be supplied and stored long-term in water.
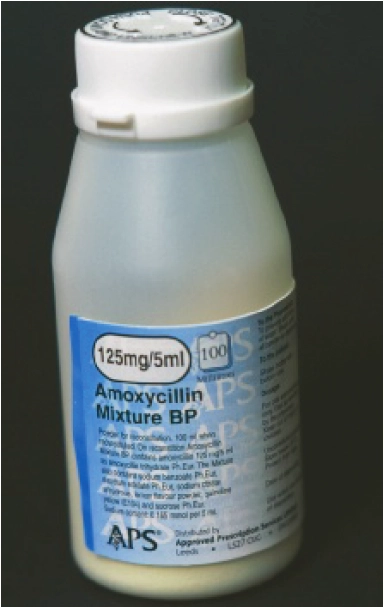
Such suspensions should also be kept in the refrigerator to minimise hydrolysis of the strained lactam ring. The b-lactam antibiotics are also susceptible to hydrolysis through the action of hydrolytic enzymes (PJ, 7/14 August 2010). Such chemical instability, and its associated implications, can have a direct effect on the chosen route of administration. One only has to look at some of the compounds that are incompatible with continuous infusion (eg, penicillins and cephalosporins), both of which contain a strained, hydrolysis-prone b-lactam ring, to understand why.
Other functional groups
Esters and amides are the most commonly encountered groups in drugs that are susceptible to hydrolysis. However, a large number of other functional groups can also react with water, resulting in broken bonds. Examples of such functional groups and the drugs in which they occur are: imines (C=N), found in diazepam; acetals (C(OR)2), found in digoxin; sulphates (ROSO3 –), found in heparin; and phosphate esters (ROPO32–), found in hydrocortisone sodium phosphate.
Preventing hydrolysis
Although hydrolysis, as outlined above, can be problematic, it can be prevented or minimised in a number of ways. For in vivo metabolism, the preventive measures are a little trickier to overcome. However, hydrolysis can be prevented by chemically modifying the structure of the active compound in the early drug development stage, providing that the problematic hydrolysis is identified early enough.
However, for inherently unstable drugs, preventive measures can include altering the dosage form in a number of ways. For liquid dosage forms, the hydrolysis of drugs is dependent on the presence of water, and thus storage of dry powders followed by reconstitution in water before dispensing minimises hydrolysis. Similarly, if a drug is known to be susceptible to hydrolysis at room temperature, its storage in a cool place is advised, and patients can be counselled on this practice. Additionally, pharmacists should provide the correct labelling on the packaging.
Moreover, the rate of hydrolysis is temperature-dependent and thus heat sterilisation of such drugs could be a problem. For semi-solid dosage forms (ointment and creams), the stability of the active ingredient can be controlled by changing the nature of the ointment or cream base in the formulation. The sensitivity of a drug to hydrolysis in a solid dosage form can be controlled in a similar manner by preparing a less hygroscopic salt of the drug or by reducing the water content in the excipients used in the formulation.
Conclusion
Pharmacists’ responsibility to provide a safe and effective product for patients requires a knowledge of the basic chemistry of drug stability. A common application in most pharmacies is consideration of drug stability in MDSs. Although definitive stability data are often lacking, guidance is available on medicines that can safely be dispensed into MDSs (PJ, 21 January 2006). Knowledge of drug chemistry will assist pharmacists’ interpretation of this, leading to improved patient safety.