

Abstract
The pharmacological management of psychotic disorders has undergone significant evolution, from early interventions with first-generation antipsychotics (FGAs) to the development of second-generation antipsychotics (SGAs) and, more recently, novel agents targeting alternative neurotransmitter systems. FGAs primarily function via dopamine D2 receptor antagonism which, despite their efficacy in alleviating positive symptoms, often leads to significant adverse effects, including extrapyramidal symptoms and metabolic disturbances. Advances in psychopharmacology have led to the introduction of SGAs, partial dopamine agonists and muscarinic receptor-targeting agents, each offering distinct mechanisms of action aimed at improving efficacy and tolerability. Emerging compounds, such as xanomeline-trospium, represent a paradigm shift by focusing on muscarinic receptor modulation, thereby avoiding dopamine receptor blockade.
This review provides a comprehensive overview of the historical progression, pharmacological mechanisms and clinical applications of antipsychotic agents, highlighting their advantages and limitations. Future research should continue to explore alternative pathways and personalised approaches to optimise treatment outcomes in psychotic disorders.
Introduction
Data suggest that psychotic disorders may affect 0.39% to 0.75% of the global population, with a higher prevalence among males and a typical onset during early adulthood1,2. These disorders significantly reduce life expectancy, largely owing to comorbidities and lifestyle factors. A nuanced understanding of the genetic and environmental factors contributing to psychotic disorders has informed the development of targeted pharmacological therapies.
The nature and diagnosis of psychosis
Psychotic disorders are characterised by profound distortions in thinking, perception and affect. The World Health Organization’s International Classification of Diseases (10th Revision) (ICD-10)2 — which is used extensively in the NHS — defines psychotic disorders under diagnoses F20 to F29, while Diagnostic and Statistical Manual of Mental Disorders (5th Edition) and International Classification of Diseases (11th RevisionI, include refinements in the classification of these conditions, emphasising dimensional and spectrum-based approaches2–4.
According to these diagnostic systems, psychotic disorders are marked by positive symptoms (e.g. hallucinations, delusions and disorganised thinking), negative symptoms (e.g. alogia, anhedonia, avolition and asociality) and cognitive impairments1. Accurate differential diagnosis relies on evaluating the duration, frequency and nature of symptoms, as well as excluding alternative causes, such as substance use, organic brain disorders and primary mood disorders2. This differentiation is likely to be critical in guiding treatment strategies.
Differential diagnosis of psychosis
The manifestations of such disorders are complex and vary widely among individuals. Differential diagnosis is often based on: the duration of an episode — acute psychotic episodes last less than one month (e.g. brief psychotic disorder), while chronic presentations persist beyond six months (e.g. schizophrenia) — the frequency of episodes, with recurrent episodes suggesting a more chronic disease; and the nature of symptoms, namely catatonia, bizarre delusions, or negative symptoms, which may help to narrow diagnoses2. This diagnostic process emphasises the need to assess symptoms holistically while considering psychosocial and medical contexts.
Aetiopathology of psychotic disorders
Genetic and environmental contributions
Psychotic disorders are likely to be highly heritable; however, most current research has focused on schizophrenia spectrum disorders. Genetic predisposition in schizophrenia-related disorders accounts for approximately 80% of the risk5. Studies on monozygotic twins demonstrate concordance rates of 40–50% and there are over 100 hundred genes involved in the pathogenesis of schizophrenia, predominantly affecting gene expression modulation, with one-third of them likely affecting glutamatergic neurotransmission6.
Key findings of the studies include associations with chromosomal deletions, such as 22q11.2, and polygenic risk scores across multiple loci7,8. Human leukocyte antigen (HLA) polymorphisms have also been implicated, with growing evidence suggesting immunological and autoimmune elements in schizophrenia9. While this represents a separate domain of study, these findings highlight the multifaceted nature of psychotic disorders. Environmental triggers often act as precipitants in genetically predisposed individuals. These include stress and bereavement, social and financial adversities, complications during labour and early-life adversities, and substance misuse and infections4.
Neurotransmitter dysregulation
The role of neurotransmitters in psychotic disorders is significant with dysregulation in dopamine, glutamate, γ-aminobutyric acid (GABA), muscarinic acetylcholine pathways and their interplay contributing to the pathophysiology of psychotic disorders10. In the dopaminergic pathways, overactivity is noted in mesolimbic pathways underlying positive symptoms, while mesocortical deficits may contribute to negative symptoms and cognitive dysfunction11,12. Most attention has been focused on the mesolimbic pathway; however, there is evidence involving the nigrostriatal pathways and the mesostriatal dopamine transmission overall, where upregulation of dopamine transmission may impact the formation of delusions — particularly persecutory ones — via dysregulation of one’s perception of the salience of environmental stimuli13. While dopamine has traditionally been considered the primary neurotransmitter implicated, advances in research suggest that glutamate, GABA and acetylcholine are also critical, particularly in specific clinical presentations. Additionally, other systems, including serotonin, adrenergic and cytokine pathways, contribute to the complexity of psychotic states14,15.
Hypofunctioning of the glutamatergic and GABA pathways has been noted, potentially involving neuronal structural abnormalities or loss16,17. N-methyl-D-aspartate receptor (NMDAR) hypofunction in the prefrontal cortex has been implicated in cognitive impairments and negative symptoms, whereas its involvement in the auditory cortex may play a pivotal role in the manifestation of hallucinations16. Similarly, hypofunction in GABAergic pathways in the prefrontal cortex and the superior temporal gyrus may be associated with impaired cognitive and social withdrawal, as well as auditory hallucinations, respectively17. Muscarinic acetylcholine receptors appear to play an orchestrating role in brain physiology by regulating the rest of the pathways mentioned18. In schizophrenia, signalling on M1 receptors, located predominantly in the frontal cortical area, and M4 receptors located in the subcortical striatal area and the hippocampus is likely downregulated, affecting cognition (i.e. M1) and signifying positive symptoms19–21.
Structural abnormalities
The interplay between neurotransmitter dysfunction and structural brain changes further compounds the pathology of psychotic disorders. Evidence suggests a 2% reduction in total brain volume, predominantly in grey matter, and a 25% enlargement of lateral ventricles, often observed in monozygotic twins discordant for schizophrenia1,22. These findings indicate not only functional dysregulation but also neurodegeneration, particularly in glutamatergic and GABAergic neurons, as described above.
Pharmacological interventions: historical and contemporary perspectives
First-generation antipsychotics (FGAs) primarily target dopamine D2 receptors and are chemically diverse, spanning phenothiazines, butyrophenones and benzamides23. Second-generation antipsychotics, such as risperidone, olanzapine and clozapine, address some of the limitations of FGAs, particularly in terms of extrapyramidal side-effects23. They offer broader symptom control and a reduced risk of extrapyramidal side effects. Agents, such as aripiprazole and cariprazine, characterised by partial D2/D3 agonism, are sometimes classified as third-generation antipsychotics owing to their unique pharmacological properties24. These agents aim to minimise cognitive and metabolic impairments associated with earlier treatments.
In 2019, Huhn et al.25 conducted a systematic review of 32 antipsychotics and highlighted that clozapine shows clear superiority over other antipsychotics, while olanzapine, risperidone, amisulpride and zotepine are potentially more effective compared to the rest. However, in the UK, there is neither a national consensus nor a specific antipsychotic treatment algorithm recommended by the National Institute for Health and Care Excellence (NICE).
Early approaches to psychosis management
The management of psychotic disorders has evolved significantly over the past two centuries, reflecting shifts in societal attitudes toward mental health and advancements in medical science. During the 19th century, psychosis treatment was predominantly rooted in the “moral treatment era”, which emphasised institutionalisation in asylums. Interventions included rudimentary and often harmful practices, such as purgatives, bleeding and ice baths, alongside more psychosocial approaches such as religious instruction and occupational therapy. Despite their limitations, these methods marked an initial attempt to address mental illness systematically26.
The early 20th century saw the advent of biological interventions, such as insulin coma therapy (introduced in 1927) and electroconvulsive therapy (ECT) in 193827,28. While these methods demonstrated some efficacy, they were often associated with severe side effects and ethical concerns. Prefrontal lobotomies, first performed in the 1930s, represented another widely used but ultimately discredited intervention29.
The emergence of modern pharmacotherapy and shifting towards community-based care
The 1950s marked a pivotal moment in psychosis treatment with the introduction of chlorpromazine, the first antipsychotic agent. Initially developed as a pre-anesthetic sedative, chlorpromazine was found to reduce psychotic symptoms significantly, transforming the landscape of mental health care30. Its discovery catalysed the transition from asylum-based care to community-based treatment, enabling many patients to lead more independent lives26.
The introduction of antipsychotic medications in the mid-20th century coincided with broader social movements advocating for deinstitutionalisation. In 1961, Enoch Powell, British minister of health (1960–1963), gave the ’Water Tower’ speech, which underscored a commitment to providing care within the community, emphasising outpatient treatment and the integration of mental health services into general healthcare settings31. This shift reflected a growing recognition of the rights of individuals with mental illness and the potential for pharmacotherapy to support community reintegration.
Development of the first antipsychotics
Chlorpromazine: the pioneer
The development of chlorpromazine, the first recognized antipsychotic, began with the synthesis of promethazine, an antihistamine with sedative properties. Promethazine’s success in managing allergies and nausea inspired further chemical modifications, leading to the development of promazine (see Figure 1).
Figure 1: chemical correlation of chlorpromazine with promazine and promethazine
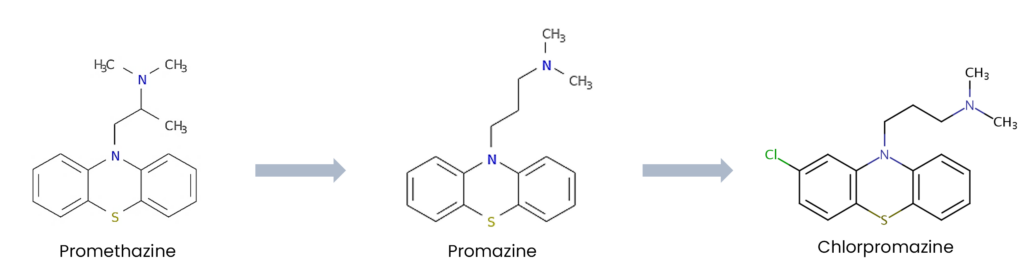
In 1950, chlorpromazine was synthesised by French chemist Paul Charpentier during his work at Rhône-Poulenc laboratories. This compound was a derivative of promazine with additional structural modifications that enhanced its central nervous system activity, leading to the introduction of phenothiazines, which is the first class of antipsychotics. Originally introduced as a pre-anesthetic agent to manage agitation in surgical patients, early clinical observations revealed its profound effects on psychotic symptoms, particularly delusions and hallucinations30. The efficacy of chlorpromazine revolutionised psychiatric care, facilitating the transition from asylum-based treatment to community-focused mental health services.
Butyrophenones and the rise of haloperidol
The butyrophenone class of antipsychotics originated in the 1950s at Janssen Pharmaceuticals during research efforts to develop stronger analgesics. Initial compounds in this class demonstrated minimal pain-relieving properties but unexpectedly exhibited pronounced effects on psychotic symptoms. Haloperidol, first synthesised in 1958, quickly gained prominence owing to its efficacy in managing paranoia and acute agitation30.
Despite sharing D2 receptor antagonism properties with chlorpromazine, haloperidol produced minimal sedation, making it particularly suitable for patients requiring clarity and alertness; however, the prevalent side effect of parkinsonism, which manifested as rigidity, tremors and bradykinesia, highlighted the limitations of early antipsychotics.
The success of haloperidol and other butyrophenones — namely droperidol and benperidol — reinforced the centrality of dopamine blockade in antipsychotic pharmacology12. However, the significant extrapyramidal side effects associated with these drugs underscored the need for advancements in drug development, ultimately paving the way for second-generation antipsychotics.
Second-generation antipsychotics: a shift towards ‘atypicality’
In the late 1950s, clozapine — a now groundbreaking antipsychotic — was developed based on the structure of imipramine, a tricyclic antidepressant, and initially failed to gain widespread acceptance owing to its unusual pharmacological profile32. Unlike other antipsychotics of the era, clozapine did not induce catalepsy or parkinsonism, leading to scepticism about its efficacy32. However, subsequent clinical trials demonstrated its superior ability to manage treatment-resistant schizophrenia, significantly reducing both positive and negative psychotic symptoms where other medications had failed, signifying clozapine as the first second-generation antipsychotics (SGA) agent33.
In 1974, the promising trajectory of clozapine was abruptly interrupted when 18 cases of severe blood dyscrasias, including agranulocytosis, were reported in Finland, resulting in 9 fatalities34. These incidents led to its withdrawal from several markets. Despite this setback, research into clozapine’s efficacy continued, and its unparalleled benefits in refractory schizophrenia led to its reapproval by the US Food and Drug Administration (FDA) in 199035. This approval came with stringent monitoring requirements, including regular full blood count (FBC) checks, to mitigate the risk of agranulocytosis.
Clozapine’s success opened the way for atypicality via the development of antipsychotic agents lacking the belonging to a specific chemical group36. Other SGAs (e.g. risperidone, olanzapine, quetiapine, aripiprazole etc.) are not specifically recommended for the treatment of treatment-resistant schizophrenia, yet they share similar lack of motor side-effect manifestation while being highly effective in treating psychotic disorders25,36.
Clozapine: a revolutionary yet complex antipsychotic
Pharmacological profile and mechanism of action of clozapine
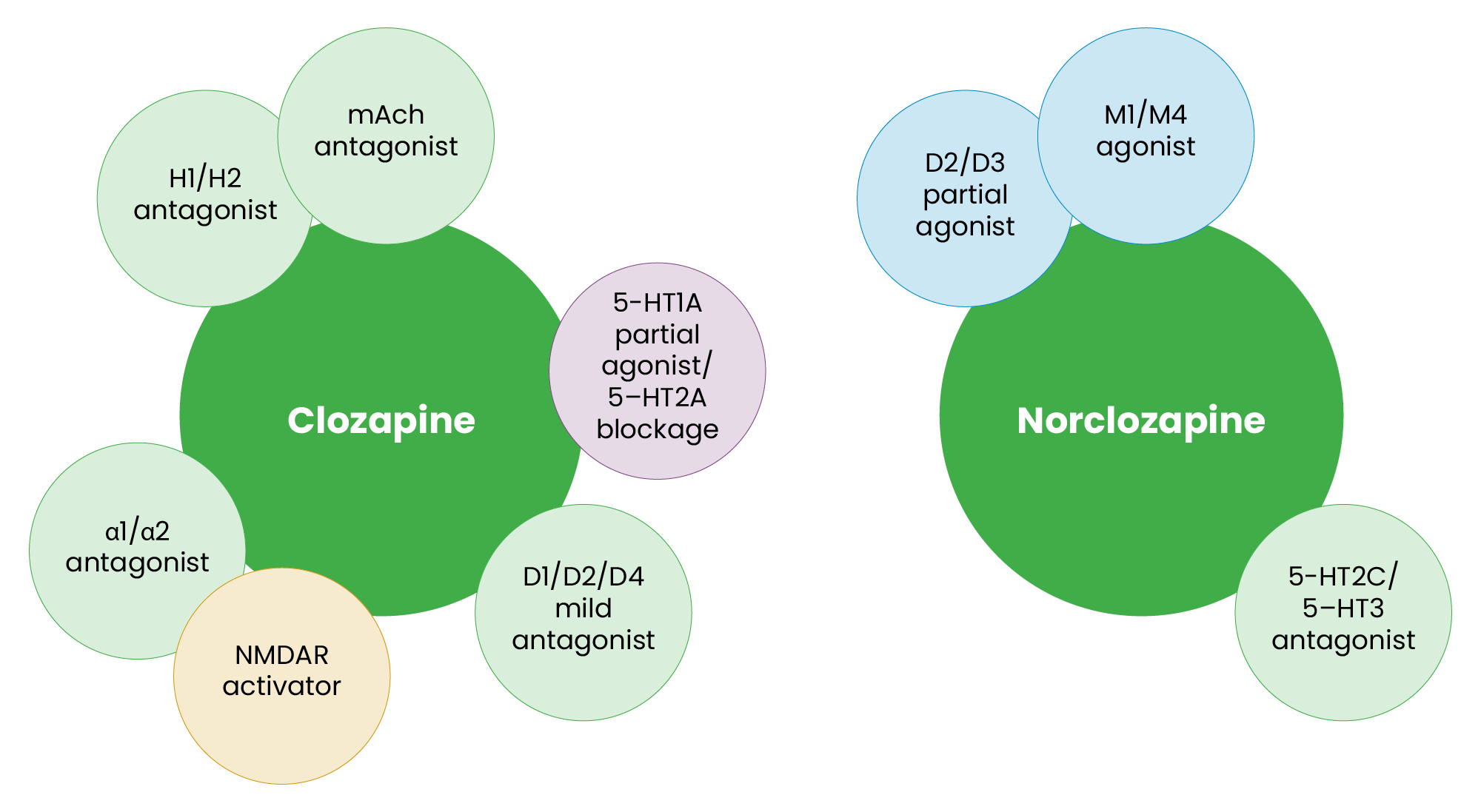
Clozapine’s unique pharmacological profile underpins its exceptional efficacy in managing schizophrenia, particularly treatment-resistant cases (see Figure 2)37. Its mechanism of action involves interactions with multiple neurotransmitter systems33. Clozapine acts as an antagonist at muscarinic receptors, while its active metabolite, norclozapine, is an agonist at M1 and M4 receptors38. Muscarinic modulation may play a pivotal role in addressing cognitive and negative symptoms of schizophrenia, aligning with the hypothesis of cholinergic dysfunction in psychosis16. It exhibits weak antagonism at D1, D2 and D4 receptors, while norclozapine functions as a partial agonist at D2 and D3 receptors, contributing to a balanced modulation of dopaminergic activity without inducing severe extrapyramidal symptoms39.
Additionally, clozapine activates N-methyl-D-aspartate receptors (NMDARs), potentially counteracting glutamate hypoactivity, which is a central feature in the pathophysiology of schizophrenia for both hallucinations and affective or cognitive elements of psychosis16,38. Its serotonergic effects include 5-HT1A partial agonism and 5-HT2A antagonism, while norclozapine antagonizes 5-HT2C and 5-HT3 receptors — interactions that are associated with improvements in mood, anxiety and psychotic symptoms39. Clozapine also antagonizes H1 and H2 histamine receptors, as well as α1 and α2 adrenergic receptors, contributing to its sedative and anxiolytic effects40.
These mechanisms align with emerging insights into the neurotransmitter dysregulation underlying psychosis. By activating NMDARs, clozapine addresses the deficits in glutamatergic signalling implicated in cognitive impairments and negative symptoms of schizophrenia16. Norclozapine’s agonism at M1 and M4 receptors mirrors the mechanism of newer antipsychotics, such as xanomeline-trospium, underscoring the relevance of muscarinic pathways in psychosis treatment19. Additionally, clozapine’s balanced dopaminergic activity reduces psychotic symptoms without the motor side effects typical of strong D212 antagonists. These interactions highlight clozapine’s ability to address multiple facets of schizophrenia, making it an invaluable tool in the psychiatric arsenal despite its challenges.
Figure 2: Pharmacological profiles of clozapine and norclozapine
Side effects and associated risks
While clozapine offers unmatched efficacy, its side-effect profile requires careful management. One of the most severe risks, agranulocytosis, is thought to be mediated by a short-lived toxic metabolite of clozapine, a nitrenium ion which is generated through the action of myeloperoxidase in the presence of H2O2 and HOCl. The nitrenium ion induces neutrophil apoptosis, leading to neutropenia41. Genetic predispositions, including certain HLA polymorphisms and variations in clozapine metabolism that increase nitrenium ion production, further elevate this risk42. Life-long FBC monitoring is mandated for all clozapine users to detect and mitigate this complication promptly. In addition to haematological concerns, clozapine’s anticholinergic activity, particularly at the M3 receptor, inhibits gastrointestinal smooth muscle contraction, delaying intestinal transit, while its antagonism of 5-HT3 receptors affects the gut-brain axis, reducing peristaltic reflexes and intraluminal secretions40. This combination can lead to severe constipation and, in extreme cases, life-threatening bowel obstruction via general gastrointestinal hypomotility, which manifests in higher prevalence compared with agranulocytosis43.
Unique mechanisms of antipsychotics with additional indications
Several antipsychotics possess unique pharmacological properties that extend beyond treating psychosis. Some of these agents are also licensed for use in depression or the management of negative symptoms in schizophrenia. The key antipsychotics in this category include quetiapine, amisulpride and flupentixol. Their distinct receptor interactions underpin their broader clinical applications.
Quetiapine: a multifaceted agent
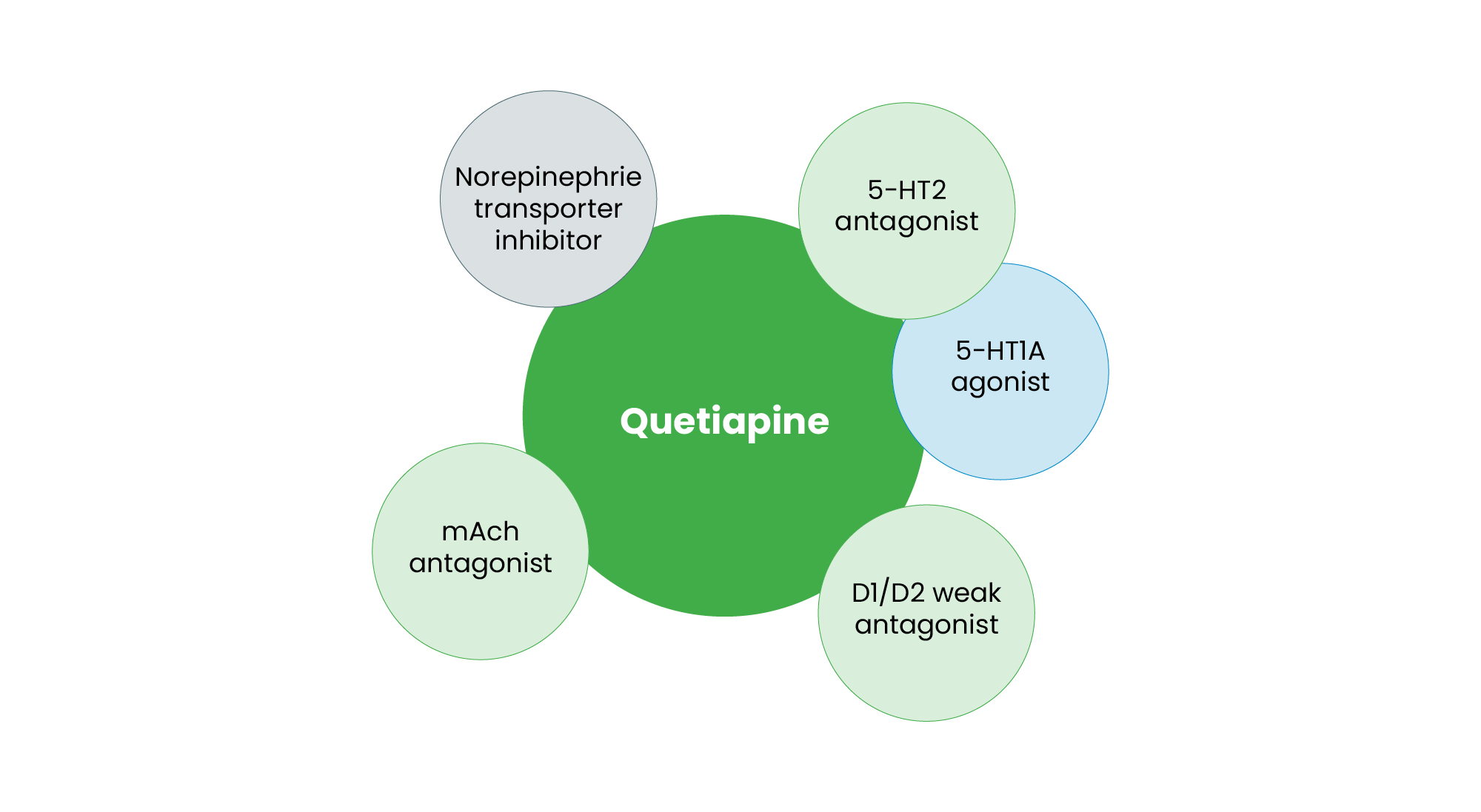
Quetiapine, an SGA, is distinguished by its affinity for multiple neurotransmitter systems (see Figure 3). It acts as a 5-HT1A agonist, which may contribute to its antidepressant properties43. Simultaneously, it functions as a 5-HT2 antagonist, a feature common among SGAs that enhances dopamine release in key cortical areas, potentially improving negative symptoms44. Quetiapine is a weak D1 and D2 antagonist, which contributes to its lower risk of extrapyramidal side effects. Additionally, its transient binding to D2 receptors results in a unique “fast-off” mechanism, reducing the likelihood of motor disturbances45. It also exhibits norepinephrine transporter (NET) inhibition, which may contribute to its mood-stabilising effects46. Furthermore, quetiapine acts as an H1 antagonist, explaining its sedative properties and its use in treating anxiety and insomnia in depressive disorders. Its broad receptor affinity contributes to its complex pharmacodynamic profile, with evidence suggesting that quetiapine’s efficacy extends beyond simple receptor antagonism to intracellular signalling modulation, neuroplasticity enhancement and effects on oxidative stress47. Owing to this complex receptor profile, quetiapine is approved for bipolar depression, major depressive disorder (MDD) (as adjunctive therapy) and schizophrenia, making it a versatile psychotropic agent46.
Figure 3: Pharmacological profile of quetiapine
Amisulpride: selective dopaminergic modulation
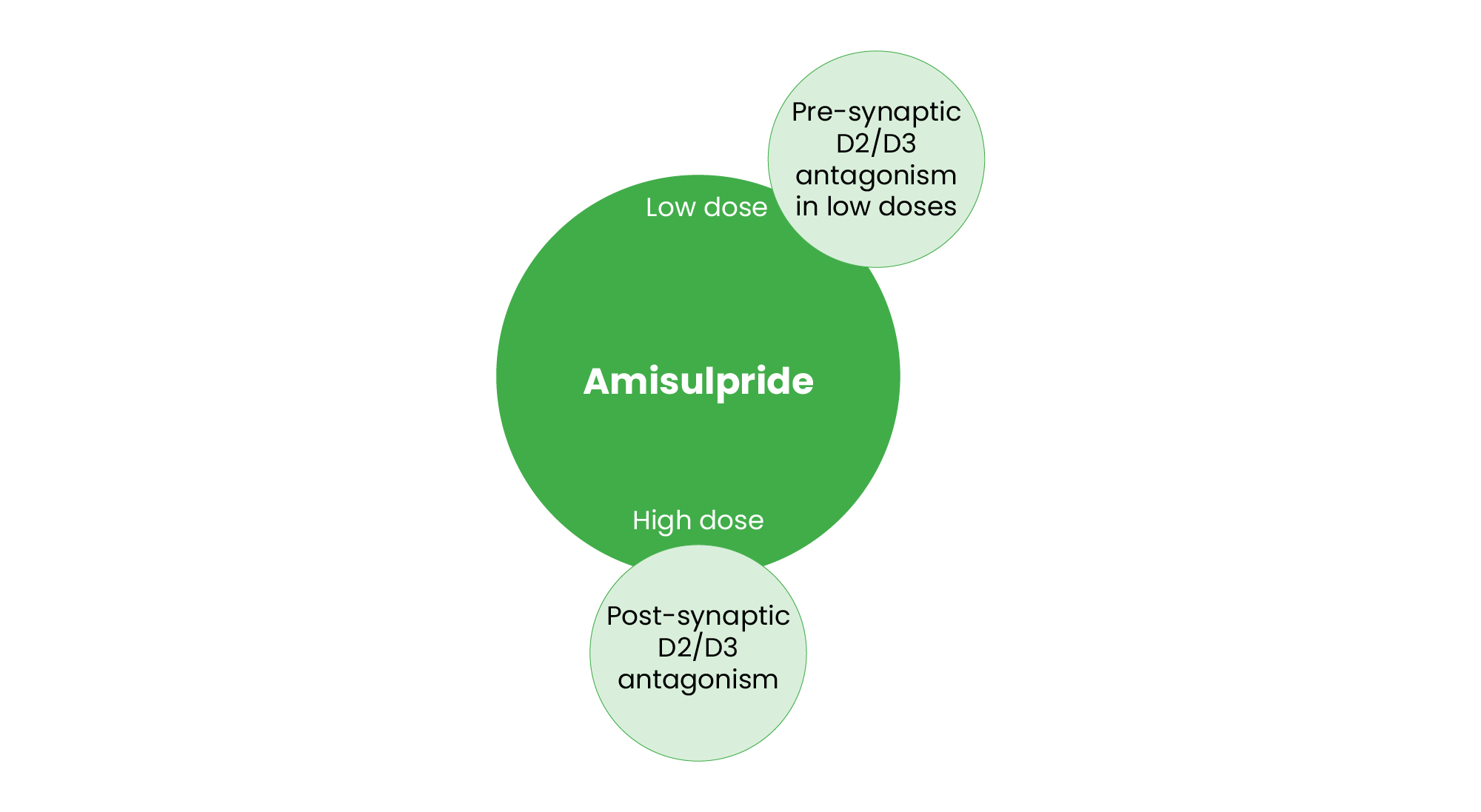
Amisulpride is unique among SGAs owing to its differential effects on dopamine receptors depending on the dosage (see Figure 4). At lower doses, amisulpride preferentially blocks presynaptic D2 and D3 receptors, leading to increased dopamine release in key brain regions48. This mechanism is believed to underlie its efficacy in alleviating negative symptoms of schizophrenia49,50. At higher doses, amisulpride functions as a postsynaptic D2 and D3 antagonist, effectively treating positive symptoms of schizophrenia while minimising extrapyramidal side effects51. Unlike other SGAs, amisulpride has negligible effects on serotonin, adrenergic and histaminergic receptors, reducing sedation and metabolic side effects52. Owing to its ability to enhance dopaminergic transmission at lower doses, amisulpride’s licence refers to patients where negative symptoms constitutes a predominant feature50. Additionally, amisulpride’s pharmacokinetics demonstrate a dose-dependent profile, with rapid absorption and a biphasic elimination pattern, contributing to its favourable tolerability in clinical use48.
Figure 4: Pharmacological profile of amisulpride
Flupentixol: outside the boundaries of psychotic disorders
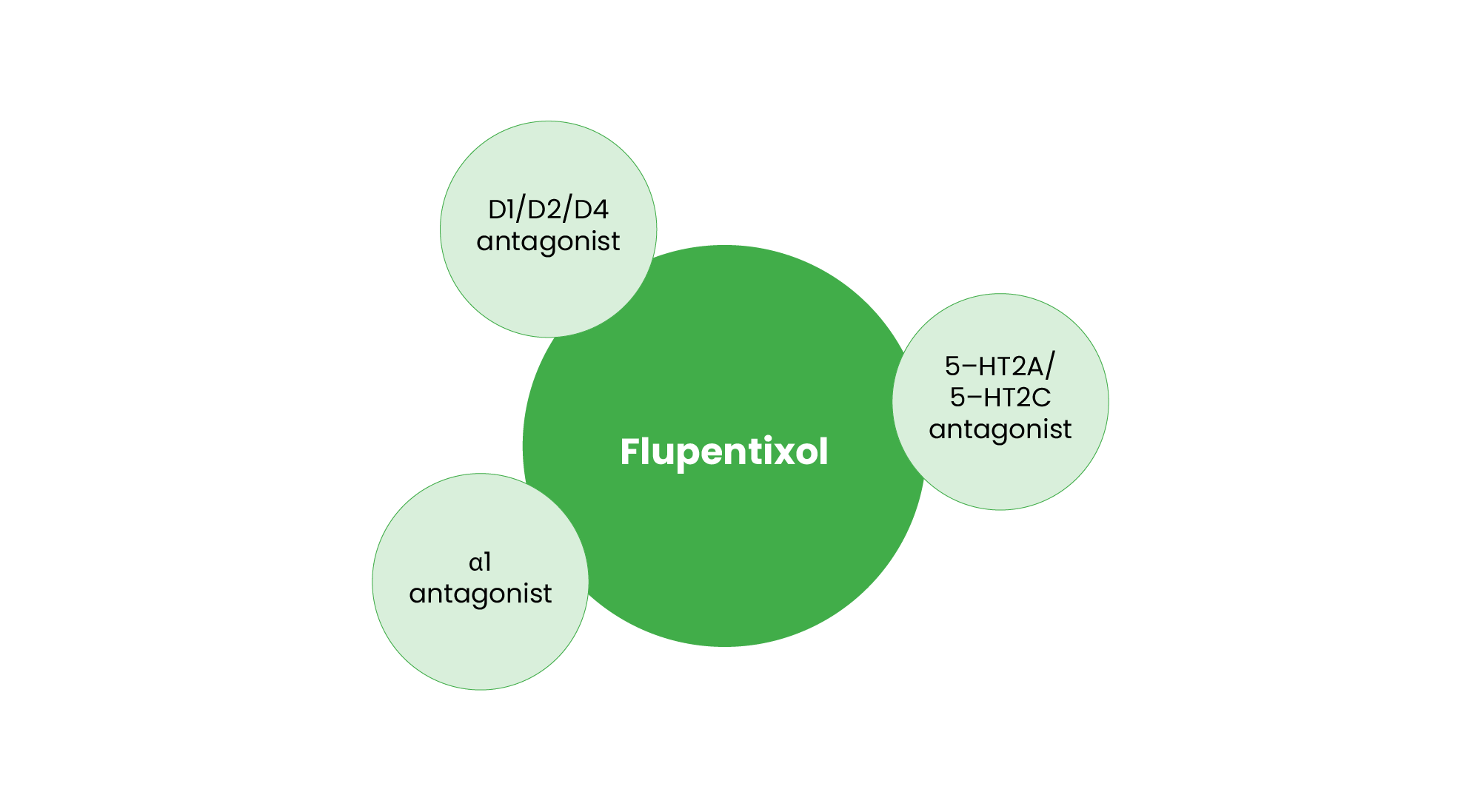
Although flupentixol is classified as an FGA, it shares some characteristics with SGAs, warranting its inclusion in this category (see Figure 5). Flupentixol is a potent D1, D2 and D3 antagonist, effectively treating positive symptoms, while it also possesses 5-HT2A and 5-HT2C antagonistic properties, along with α1 adrenergic antagonism, which contribute to its antidepressant effects53. Clinically, flupentixol is licensed in some countries for mild-to-moderate depression in low doses, particularly in cases where patients exhibit apathy and reduced motivation4.
The unique receptor interactions of these SGAs enable their expanded use beyond psychosis treatment. Quetiapine’s serotonergic and noradrenergic effects support its antidepressant action, amisulpride’s selective dopaminergic modulation targets negative symptoms, and flupentixol’s serotonergic and noradrenergic activity broadens its clinical utility. These pharmacological distinctions underscore the evolving role of antipsychotics in psychiatric care, facilitating personalised treatment approaches based on symptomatology and patient needs.
Figure 5: Pharmacological profile of flupentixol
Partial agonists at D2 and D3 receptors: a shift in antipsychotic strategy
The development of partial dopamine D2 and D3 receptor agonists emerged in the early 2000s as an attempt to address the limitations of older SGAs. While most SGAs act as full antagonists at dopamine receptors, this approach has been associated with cognitive impairment and significant metabolic side effects, including weight gain and dyslipidaemia54. Partial agonists offer a more balanced approach by stabilising dopaminergic signalling rather than fully blocking it, thereby mitigating these adverse effects55.
Aripiprazole was the first of this class and was introduced as a dopamine-serotonin system stabiliser. By acting as a partial agonist at D2 and D3 receptors, it modulates dopamine activity based on endogenous tone, enhancing signalling in deficient states while dampening excessive transmission. This mechanism reduces extrapyramidal symptoms and metabolic burden compared with traditional SGAs56. Aripiprazole has been widely used off label for behavioural issues in autism spectrum disorder (ASD), obsessive-compulsive disorder (OCD) and tic disorders57. Cariprazine, with a higher affinity for D3 receptors compared with aripiprazole, has been proposed to offer enhanced efficacy in addressing negative symptoms of schizophrenia58. This distinct receptor profile may contribute to improved cognitive function and motivational deficits, which are often unresponsive to standard dopamine antagonists. Similarly, brexpiprazole acts as a partial agonist at D2 and D3 receptors but has additional serotonin receptor activity, particularly at 5-HT1A and 5-HT2A sites, which may contribute to its anxiolytic and antidepressant effects59. It has been explored as an adjunctive therapy in MDD and is often used to counterbalance the metabolic risks associated with other antipsychotics.
Such agents, namely aripiprazole, are frequently prescribed alongside primary antipsychotic treatments to offset metabolic side effects60. While they may reduce weight gain and insulin resistance associated with conventional SGAs, there is speculation that their partial agonist activity could compromise the full antipsychotic efficacy of the primary medication, particularly in treatment-resistant schizophrenia61. Regardless, the introduction of partial dopamine agonists represents a paradigm shift in antipsychotic therapy, offering potential advantages in cognitive preservation and metabolic risk reduction. While their use extends beyond psychotic disorders to conditions such as ASD, OCD and tic disorders, ongoing research is required to optimise their role in adjunctive therapy and determine their long-term benefits and risks.
Emerging antipsychotic agents: pimavanserin and lumateperone
Pimavanserin: a focus on serotoninergic pathways
Pimavanserin, approved by the FDA in 2016, represents a breakthrough in the treatment of psychosis, particularly Parkinson’s disease psychosis62. Unlike first-generation and many other antipsychotics, which primarily target dopamine receptors, pimavanserin functions as an inverse agonist and antagonist at 5-HT2A receptors with minimal affinity for dopaminergic receptors63. This mechanism allows for the reduction of psychotic symptoms without exacerbating motor dysfunction, which is a significant concern in patients with Parkinson’s disease64. Additionally, there is emerging interest in the potential application of pimavanserin for schizophrenia-related psychosis, particularly in individuals with prominent negative symptoms or treatment resistance65.
Lumateperone: an intentionally multi-targeted mechanism
Lumateperone, approved in 2019, offers a novel pharmacological profile that differentiates it from other SGAs. It simultaneously modulates dopamine, serotonin and glutamate systems, contributing to its broad efficacy and improved tolerability66. Lumateperone acts as a dopamine D2 receptor modulator, a 5-HT2A antagonist and enhances glutamatergic signalling via NMDA receptor modulation67. This combined activity is believed to contribute to its efficacy in treating schizophrenia while reducing the risk of extrapyramidal symptoms and metabolic side effects. The results of clinical trials, published in 2023, have demonstrated its ability to improve both positive and negative symptoms with a favourable safety profile compared to older antipsychotics68.
Pimavanserin and lumateperone represent significant advancements in antipsychotic therapy by offering targeted mechanisms of action with improved safety profiles. Pimavanserin’s selective 5-HT2A antagonism makes it particularly useful in Parkinson’s disease psychosis, while lumateperone’s modulation of multiple neurotransmitter systems provides a promising alternative for schizophrenia treatment. These agents underscore the ongoing efforts to refine antipsychotic therapy, aiming to enhance efficacy while minimising side effects.
Xanomeline–trospium: a new frontier in schizophrenia treatment
In September 2024, the FDA approved the xanomeline-trospium combination, a novel antipsychotic that represents a significant shift in the pharmacological management of schizophrenia. Currently, this combination is marketed under the brand name Cobenfy (Bristol Myers Squibb)69. Unlike other antipsychotics, which primarily target dopamine and serotonin receptors, xanomeline acts as an agonist at muscarinic M1 and M4 receptors. This mechanism is particularly relevant given the growing evidence supporting the role of cholinergic dysfunction in schizophrenia, especially in cognitive and negative symptom domains19,70.
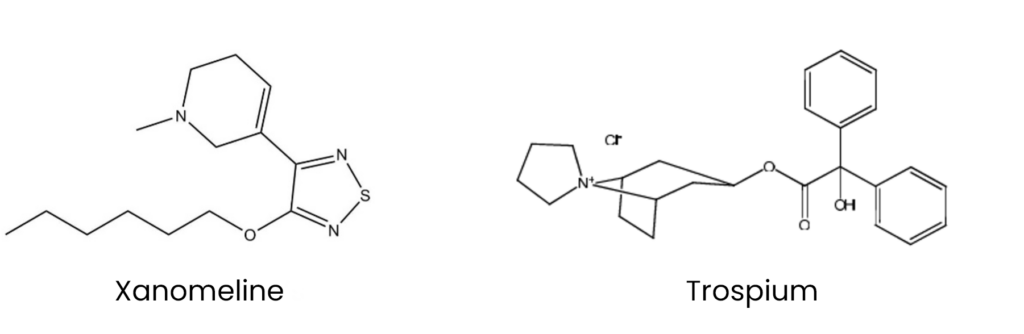
Xanomeline was initially developed in the 1990s and trialled as a potential treatment for Alzheimer’s disease (see Figure 6)69. Despite demonstrating pro-cognitive effects, its clinical development was halted owing to dose-limiting peripheral cholinergic side effects71,72. Trospium, a quaternary ammonium compound first synthesised in 1967, was later incorporated to mitigate these adverse effects (see Figure 7). It acts as a peripherally restricted muscarinic antagonist and was first approved for medical use in Germany in 1999, followed by FDA approval in 2004 and MHRA approval in 2008 for the symptomatic treatment of overactive bladder73,74.
Figure 6 and 7: Chemical structures of xanomeline and trospium
To mitigate peripheral cholinergic side effects, it is co-formulated with trospium, a peripherally restricted muscarinic antagonist and marketed in the UK as a standalone symptomatic treatment of overactive bladder73,74. The M1 receptor plays a crucial role in modulating cognitive function, synaptic plasticity and neuronal excitability. Its activation is hypothesised to improve cognitive deficits observed in schizophrenia, which are often resistant to standard antipsychotic treatments19. Additionally, the M4 receptor modulates dopamine release, particularly in the striatum. By activating M4 receptors, xanomeline indirectly reduces excessive dopaminergic signalling, contributing to the attenuation of psychotic symptoms without the need for direct D2 receptor blockade. This mechanism is expected to lower the risk of extrapyramidal side effects commonly associated with dopamine antagonists71.
Xanomeline’s novel mechanism offers several potential benefits. Unlike conventional antipsychotics, which often exacerbate cognitive impairment, M1 agonism may enhance cognitive function and executive processing75,76. By avoiding direct dopamine D2 receptor antagonism, xanomeline is less likely to induce movement disorders such as parkinsonism and tardive dyskinesia. Additionally, M4 receptor activity has been linked to improved motivational and social engagement behaviours, addressing a significant unmet need in schizophrenia treatment19,77.
Preliminary clinical trials have demonstrated promising efficacy, with significant reductions in both positive and negative symptoms of schizophrenia78. Compared with SGAs, xanomeline-trospium exhibited comparable efficacy in treating hallucinations and delusions, superior improvements in cognitive performance, and a lower incidence of weight gain and metabolic disturbances78,79. The approval of Cobenfy marks a new frontier in antipsychotic development. By shifting the focus from dopaminergic antagonism to cholinergic modulation, this drug offers an innovative treatment pathway with the potential to address challenges around antipsychotic-induced side-effects, directly linking to patient satisfaction and adherence. As further post-market studies emerge, its long-term impact on clinical outcomes and real-world effectiveness will become clearer, potentially reshaping the future of psychopharmacology.
Discussion and conclusion
The evolution of antipsychotic treatment reflects ongoing attempts to balance efficacy with tolerability. FGAs demonstrated clear effectiveness for positive symptoms through dopamine D2 antagonism but were limited by extrapyramidal adverse effects. Second-generation agents improved tolerability and expanded symptom coverage yet introduced significant metabolic burden and offered limited benefit for negative and cognitive symptoms. These persistent shortcomings have driven the development of newer treatments, including dopamine partial agonists and non-dopaminergic agents, marking a shift towards more targeted and mechanistically informed pharmacotherapy. The introduction of dopamine partial agonists, such as aripiprazole and cariprazine, aimed to modulate dopaminergic activity more precisely, reducing the burden of side effects while maintaining therapeutic efficacy.
Recent advancements have moved us from a dopamine-centred approach to the development of receptor-specific agents, such as pimavanserin, for Parkinson’s disease psychosis and lumateperone for schizophrenia, which underscores the shift towards more targeted pharmacotherapy with potential benefits to users.
Perhaps the most significant advancement in recent years is the approval of xanomeline-trospium. By selectively targeting muscarinic acetylcholine receptors, the xanomeline-trospium bitherapy offers a novel mechanism that could address both cognitive and negative symptoms of schizophrenia while reducing the risk of motor and metabolic side effects. This highlights a broader trend towards developing treatments that align more closely with the neurobiological underpinnings of psychosis.
Despite these advances, clozapine remains the most effective antipsychotic, being the only antipsychotic with proven efficacy in treatment-resistant schizophrenia. Its complex and elusive mechanism of action involves multiple neurotransmitter systems, including muscarinic receptor modulation (M1 and M4) and NMDAR activation, as well as effects on serotonin and adrenergic receptors. This diverse pharmacological profile is shared with some emerging agents, such as xanomeline-trospium and lumateperone, reinforcing the relevance of these pathways in psychosis treatment. Clozapine’s unique effectiveness highlights the need for further research into muscarinic and glutamatergic modulation as potential avenues for developing next-generation antipsychotics. Understanding how these mechanisms contribute to clozapine’s superior efficacy may drive innovation in antipsychotic drug development.
The heterogeneity of psychotic disorders necessitates personalised treatment strategies, yet current pharmacological options often rely on a trial-and-error approach. Further research is needed to identify predictive biomarkers that can guide treatment selection and improve patient outcomes. Additionally, long-term studies are required to fully assess the safety and efficacy of novel agents such as xanomeline-trospium in real-world clinical settings.
In conclusion, while substantial progress has been made in the pharmacological management of psychotic disorders, ongoing research into alternative mechanisms of action, individualised treatment approaches and long-term safety data will be critical in shaping the future of psychiatric care. The integration of novel therapies into clinical practice must be accompanied by continued investigation into their impact on both symptoms and overall patient wellbeing.
- 1.Moreno-Küstner B, Martín C, Pastor L. Prevalence of psychotic disorders and its association with methodological issues. A systematic review and meta-analyses. McKenna PJ, ed. PLoS ONE. 2018;13(4):e0195687. doi:10.1371/journal.pone.0195687
- 2.International statistical classification of diseases and related health problems (11th ed.). World Health Organization. 2019. https://www.who.int/standards/classifications/classification-of-diseases
- 3.Psychosis and schizophrenia in adults: treatment and management (NICE guideline CG178). National Institute for Health and Care Excellence . 2014. https://www.nice.org.uk/guidance/cg178
- 4.Depression in adults: Recognition and management. National Institute for Health and Care Excellence . 2022. https://www.nice.org.uk/guidance/ng222
- 5.Marder SR, Cannon TD. Schizophrenia. Ropper AH, ed. N Engl J Med. 2019;381(18):1753-1761. doi:10.1056/nejmra1808803
- 6.Coyle JT, Ruzicka WB, Balu DT. Fifty Years of Research on Schizophrenia: The Ascendance of the Glutamatergic Synapse. AJP. 2020;177(12):1119-1128. doi:10.1176/appi.ajp.2020.20101481
- 7.Bassett AS. Chromosomal Aberrations and Schizophrenia. Br J Psychiatry. 1992;161(3):323-334. doi:10.1192/bjp.161.3.323
- 8.Owen MJ, Craddock N, Jablensky A. The Genetic Deconstruction of Psychosis. Schizophrenia Bulletin. 2007;33(4):905-911. doi:10.1093/schbul/sbm053
- 9.Tamouza R, Krishnamoorthy R, Leboyer M. Understanding the genetic contribution of the human leukocyte antigen system to common major psychiatric disorders in a world pandemic context. Brain, Behavior, and Immunity. 2021;91:731-739. doi:10.1016/j.bbi.2020.09.033
- 10.Mlambo R, Liu J, Wang Q, Tan S, Chen C. Receptors Involved in Mental Disorders and the Use of Clozapine, Chlorpromazine, Olanzapine, and Aripiprazole to Treat Mental Disorders. Pharmaceuticals. 2023;16(4):603. doi:10.3390/ph16040603
- 11.Orsolini L, Pompili S, Volpe U. Schizophrenia: A Narrative Review of Etiopathogenetic, Diagnostic and Treatment Aspects. JCM. 2022;11(17):5040. doi:10.3390/jcm11175040
- 12.Gründer G, Cumming P. The Dopamine Hypothesis of Schizophrenia. The Neurobiology of Schizophrenia. Published online 2016:109-124. doi:10.1016/b978-0-12-801829-3.00015-x
- 13.McCutcheon RA, Krystal JH, Howes OD. Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World Psychiatry. 2020;19(1):15-33. doi:10.1002/wps.20693
- 14.Stahl SM. Beyond the dopamine hypothesis of schizophrenia to three neural networks of psychosis: dopamine, serotonin, and glutamate. CNS Spectr. 2018;23(3):187-191. doi:10.1017/s1092852918001013
- 15.Kim H, Baek SH, Kim JW, et al. Inflammatory markers of symptomatic remission at 6 months in patients with first-episode schizophrenia. Schizophr. 2023;9(1). doi:10.1038/s41537-023-00398-1
- 16.Kruse AO, Bustillo JR. Glutamatergic dysfunction in Schizophrenia. Transl Psychiatry. 2022;12(1). doi:10.1038/s41398-022-02253-w
- 17.de Jonge JC, Vinkers CH, Hulshoff Pol HE, Marsman A. GABAergic Mechanisms in Schizophrenia: Linking Postmortem and In Vivo Studies. Front Psychiatry. 2017;8. doi:10.3389/fpsyt.2017.00118
- 18.Singh T, Sharma S, Dhiman S. Neurotransmitter Systems and the Nicotine Dependence-Induced Withdrawal Syndrome. Neuropathology of Drug Addictions and Substance Misuse . Published online 2016.
- 19.Dean B, Bakker G, Ueda HR, Tobin AB, Brown A, Kanaan RAA. A growing understanding of the role of muscarinic receptors in the molecular pathology and treatment of schizophrenia. Front Cell Neurosci. 2023;17. doi:10.3389/fncel.2023.1124333
- 20.Jones SE, Harvey PD. Cross-diagnostic determinants of cognitive functioning: the muscarinic cholinergic receptor as a model system. Transl Psychiatry. 2023;13(1). doi:10.1038/s41398-023-02400-x
- 21.Yohn SE, Weiden PJ, Felder CC, Stahl SM. Muscarinic acetylcholine receptors for psychotic disorders: bench-side to clinic. Trends in Pharmacological Sciences. 2022;43(12):1098-1112. doi:10.1016/j.tips.2022.09.006
- 22.Woolley J, McGuire P. Neuroimaging in schizophrenia: what does it tell the clinician? Adv psychiatr treat. 2005;11(3):195-202. doi:10.1192/apt.11.3.195
- 23.Chokhawala K, Stevens L. Antipsychotic Medications. StatPearls ; 2023.
- 24.Mailman R, Murthy V. Third Generation Antipsychotic Drugs: Partial Agonism or Receptor Functional Selectivity? CPD. 2010;16(5):488-501. doi:10.2174/138161210790361461
- 25.Huhn M, Nikolakopoulou A, Schneider-Thoma J, et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: a systematic review and network meta-analysis. The Lancet. 2019;394(10202):939-951. doi:10.1016/s0140-6736(19)31135-3
- 26.Taubes T. “Healthy Avenues of the Mind”: Psychological Theory Building and the Influence of Religion During the Era of Moral Treatment. AJP. 1998;155(8):1001-1008. doi:10.1176/ajp.155.8.1001
- 27.Doroshow DB. Performing a Cure for Schizophrenia: Insulin Coma Therapy on the Wards. Journal of the History of Medicine and Allied Sciences. 2006;62(2):213-243. doi:10.1093/jhmas/jrl044
- 28.Gazdag G, Ungvari GS. Electroconvulsive therapy: 80 years old and still going strong. WJP. 2019;9(1):1-6. doi:10.5498/wjp.v9.i1.1
- 29.Fleming GWTH. An Introduction to Physical Methods of Treatment in Psychiatry. By William Sargant and Eliot Slater. Edinburgh: E. & S. Livingstone, 1948. Price 10s. 6d. J ment sci. 1949;95(401):991-992. doi:10.1192/bjp.95.401.991-a
- 30.Ramachandraiah C, Subramaniam N, Tancer M. The story of antipsychotics: Past and present. Indian J Psychiatry. 2009;51(4):324. doi:10.4103/0019-5545.58304
- 31.Gilburt H, Peck A, Edwards N, Naylor C. Service transformation: Lessons from mental health. Kings Fund . 2014. https://assets.kingsfund.org.uk/f/256914/x/88feefd75c/service_transformation_february_2014.pdf
- 32.Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60 Suppl 12:22-23. https://www.ncbi.nlm.nih.gov/pubmed/10372606
- 33.Kane J. Clozapine for the Treatment-Resistant Schizophrenic. Arch Gen Psychiatry. 1988;45(9):789. doi:10.1001/archpsyc.1988.01800330013001
- 34.Idänpään-Heikkilä J, Alhava E, Olkinuora M, Palva I. CLOZAPINE AND AGRANULOCYTOSIS. The Lancet. 1975;306(7935):611. doi:10.1016/s0140-6736(75)90206-8
- 35.Crilly J. The history of clozapine and its emergence in the US market. Hist Psychiatry. 2007;18(1):39-60. doi:10.1177/0957154×07070335
- 36.Lyman M, McCutcheon RA. Antipsychotic drugs at 75: the past, present, and future of psychosis management. British Medical Bulletin. 2025;156(1). doi:10.1093/bmb/ldaf016
- 37.Haidary H, Padhy R. Clozapine. StatPearls; 2023.
- 38.Wenthur CJ, Lindsley CW. Classics in Chemical Neuroscience: Clozapine. ACS Chem Neurosci. 2013;4(7):1018-1025. doi:10.1021/cn400121z
- 39.Dell’Osso L, Bonelli C, Nardi B, et al. Rethinking Clozapine: Lights and Shadows of a Revolutionary Drug. Brain Sciences. 2024;14(1):103. doi:10.3390/brainsci14010103
- 40.Shirazi A, Stubbs B, Gomez L, et al. Prevalence and Predictors of Clozapine-Associated Constipation: A Systematic Review and Meta-Analysis. IJMS. 2016;17(6):863. doi:10.3390/ijms17060863
- 41.Ramli FF, Ali A, Syed Hashim SA, Kamisah Y, Ibrahim N. Reduction in Absolute Neutrophil Counts in Patient on Clozapine Infected with COVID-19. IJERPH. 2021;18(21):11289. doi:10.3390/ijerph182111289
- 42.Mijovic A, MacCabe JH. Clozapine-induced agranulocytosis. Ann Hematol. 2020;99(11):2477-2482. doi:10.1007/s00277-020-04215-y
- 43.Cross AJ, Widzowski D, Maciag C, et al. Quetiapine and its metabolite norquetiapine: translation from in vitro pharmacology to in vivo efficacy in rodent models. British J Pharmacology. 2015;173(1):155-166. doi:10.1111/bph.13346
- 44.Maan J, Ershadi M, Saadabadi A. Quetiapine. StatPearls; 2023.
- 45.Seeman P. Dopamine D2 receptors as treatment targets in schizophrenia. Clin Schizophr Relat Psychoses. 2010;4(1):56-73. https://www.ncbi.nlm.nih.gov/pubmed/20643630
- 46.Riedel M, Müller N, Strassnig M, Spellmann I, Severus E, Möller HJ. Quetiapine in the treatment of schizophrenia and related disorders. Neuropsychiatric Disease and Treatment. 2007;3(2):219-235. doi:10.2147/nedt.2007.3.2.219
- 47.Ignácio ZM, Calixto AV, da Silva RH, Quevedo J, Réus GZ. The use of quetiapine in the treatment of major depressive disorder: Evidence from clinical and experimental studies. Neuroscience & Biobehavioral Reviews. 2018;86:36-50. doi:10.1016/j.neubiorev.2017.12.012
- 48.Rosenzweig P, Canal M, Patat A, Bergougnan L, Zieleniuk I, Bianchetti G. A review of the pharmacokinetics, tolerability and pharmacodynamics of amisulpride in healthy volunteers. Human Psychopharmacology. 2002;17(1):1-13. doi:10.1002/hup.320
- 49.Nielsen MØ, Kristensen TD, Borup Bojesen K, Glenthøj BY, Lemvigh CK, Ebdrup BH. Differential Effects of Aripiprazole and Amisulpride on Negative and Cognitive Symptoms in Patients With First-Episode Psychoses. Front Psychiatry. 2022;13. doi:10.3389/fpsyt.2022.834333
- 50.Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. The Lancet. 2009;373(9657):31-41. doi:10.1016/s0140-6736(08)61764-x
- 51.Möller H. Amisulpride: a review of its efficacy in schizophrenia. Acta Psychiatr Scand Suppl. 2000;400:17-22. https://www.ncbi.nlm.nih.gov/pubmed/10823307
- 52.Schoemaker H, Claustre Y, Fage D, et al. Neurochemical characteristics of amisulpride, an atypical dopamine D2/D3 receptor antagonist with both presynaptic and limbic selectivity. J Pharmacol Exp Ther. 1997;280(1):83-97. https://www.ncbi.nlm.nih.gov/pubmed/8996185
- 53.Mahapatra J, Quraishi SN, David A, Sampson S, Adams CE. Flupenthixol decanoate (depot) for schizophrenia or other similar psychotic disorders. Cochrane Database of Systematic Reviews. 2014;2014(6). doi:10.1002/14651858.cd001470.pub2
- 54.Kane JM, Correll CU. Optimizing Treatment Choices to Improve Adherence and Outcomes in Schizophrenia. J Clin Psychiatry. 2019;80(5). doi:10.4088/jcp.in18031ah1c
- 55.Cookson J, Pimm J, Reynolds G. Partial agonists of dopamine receptors: clinical effects and dopamine receptor interactions in combining aripiprazole with a full antagonist in treating psychosis. NHS. 2023. https://www.elft.nhs.uk/research/resource/partial-agonists-dopamine-receptors-clinical-effects-and-dopamine-receptor
- 56.Stahl S. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. 4th ed. Cambridge University; 2013. https://assets.cambridge.org/97811070/25981/frontmatter/9781107025981_frontmatter.pdf
- 57.Hollander E, Phillips A, Yeh C. Targeted treatments for symptom domains in child and adolescent autism. Lancet. Published online 2003.
- 58.Correll CU, Schooler NR. <p>Negative Symptoms in Schizophrenia: A Review and Clinical Guide for Recognition, Assessment, and Treatment</p> NDT. 2020;Volume 16:519-534. doi:10.2147/ndt.s225643
- 59.Diefenderfer LA, Iuppa C. Brexpiprazole: A review of a new treatment option for schizophrenia and major depressive disorder. Mental Health Clinician. 2017;7(5):207-212. doi:10.9740/mhc.2017.09.207
- 60.Simmons S, Sadiq S. The Impact of Adjunctive Aripiprazole on Olanzapine‐Induced Metabolic Adverse Effects in Patients With Schizophrenia: A Systematic Review. Neuropsychopharm Rep. 2025;45(3). doi:10.1002/npr2.70046
- 61.Correll CU, Rubio JM, Kane JM. What is the risk‐benefit ratio of long‐term antipsychotic treatment in people with schizophrenia? World Psychiatry. 2018;17(2):149-160. doi:10.1002/wps.20516
- 62.Cusick E, Gupta V. Pimavanserin. StatPearls; 2023.
- 63.Meltzer HY, Mills R, Revell S, et al. Pimavanserin, a Serotonin2A Receptor Inverse Agonist, for the Treatment of Parkinson’s Disease Psychosis. Neuropsychopharmacol. 2009;35(4):881-892. doi:10.1038/npp.2009.176
- 64.Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. The Lancet. 2014;383(9916):533-540. doi:10.1016/s0140-6736(13)62106-6
- 65.Ballard C, Creese B, Corbett A, Aarsland D. Atypical antipsychotics for the treatment of behavioral and psychological symptoms in dementia, with a particular focus on longer term outcomes and mortality. Expert Opinion on Drug Safety. 2010;10(1):35-43. doi:10.1517/14740338.2010.506711
- 66.Cooper D, Gupta V. Lumateperone. StatPearls; 2023.
- 67.Correll CU, Davis RE, Weingart M, et al. Efficacy and Safety of Lumateperone for Treatment of Schizophrenia. JAMA Psychiatry. 2020;77(4):349. doi:10.1001/jamapsychiatry.2019.4379
- 68.Longo G, Cicolini A, Orsolini L, Volpe U. The Novel Antipsychotic Lumateperone (Iti-007) in the Treatment of Schizophrenia: A Systematic Review. Brain Sciences. 2023;13(12):1641. doi:10.3390/brainsci13121641
- 69.Cobenfy® (Xanomeline-Trospium) Prescribing Information. Bristol Myers Squibb. 2025. https://packageinserts.bms.com/pi/pi_cobenfy.pdf
- 70.Krystal JH, Murray JD, Chekroud AM, et al. Computational Psychiatry and the Challenge of Schizophrenia. Schizophrenia Bulletin. 2017;43(3):473-475. doi:10.1093/schbul/sbx025
- 71.Bender AM, Jones CK, Lindsley CW. Classics in Chemical Neuroscience: Xanomeline. ACS Chem Neurosci. 2017;8(3):435-443. doi:10.1021/acschemneuro.7b00001
- 72.Kidambi N, Elsayed OH, El-Mallakh RS. Xanomeline-Trospium and Muscarinic Involvement in Schizophrenia. NDT. 2023;Volume 19:1145-1151. doi:10.2147/ndt.s406371
- 73.Compound Summary for CID 5284632, Trospium. PubChem. 2025. https://pubchem.ncbi.nlm.nih.gov/compound/Trospium
- 74.Regurin XL 60mg (trospium chloride): Summary of Product Characteristics. . EMC. 2025. https://www.medicines.org.uk/emc/product/6644/smpc#gref
- 75.Foster DJ, Bryant ZK, Conn PJ. Targeting muscarinic receptors to treat schizophrenia. Behavioural Brain Research. 2021;405:113201. doi:10.1016/j.bbr.2021.113201
- 76.Paul SM, Yohn SE, Popiolek M, Miller AC, Felder CC. Muscarinic Acetylcholine Receptor Agonists as Novel Treatments for Schizophrenia. AJP. 2022;179(9):611-627. doi:10.1176/appi.ajp.21101083
- 77.Yohn SE, Harvey PD, Brannan SK, Horan WP. The potential of muscarinic M1 and M4 receptor activators for the treatment of cognitive impairment associated with schizophrenia. Front Psychiatry. 2024;15. doi:10.3389/fpsyt.2024.1421554
- 78.Kaul I, Sawchak S, Walling DP, et al. Efficacy and Safety of Xanomeline-Trospium Chloride in Schizophrenia. JAMA Psychiatry. 2024;81(8):749. doi:10.1001/jamapsychiatry.2024.0785
- 79.Kaul I, Sawchak S, Correll CU, et al. Efficacy and safety of the muscarinic receptor agonist KarXT (xanomeline–trospium) in schizophrenia (EMERGENT-2) in the USA: results from a randomised, double-blind, placebo-controlled, flexible-dose phase 3 trial. The Lancet. 2024;403(10422):160-170. doi:10.1016/s0140-6736(23)02190-6


