Key points:
- Chronic hepatitis B virus (HBV) and C virus (HCV) infections are the leading cause of hepatocellular carcinoma (HCC) worldwide.
- Functional cure of HBV consists of hepatitis B surface antigen (HBsAg) loss with or without hepatitis B surface antibody (HBsAb) seroconversion, with undetectable serum HBV DNA. cccDNA still persists in functional cure but it is transcriptionally inactive. Complete cure of HBV requires permanent silencing or elimination of covalently closed circular DNA (cccDNA).
- While current nucleos(t)ide analogue therapy for chronic HBV reduces fibrosis and the incidence of HCC, it rarely leads to a functional cure.
- Novel therapies have been developed for chronic HBV that inhibit viral entry into hepatocyte, nucleocapsid and viral assembly, target cccDNA, prevent HBsAg secretion and restore host adaptive and innate immunity.
- The development of direct-acting antivirals (DAAs) has led to cure rates exceeding 90% in many populations.
- Effective therapy for chronic HCV now exists for all patient populations including patients with decompensated cirrhosis, HIV co-infection and chronic kidney disease (CKD).
- The next frontier of chronic HCV therapy will involve pan-genotypic regimens, likely with a shorter duration.
Introduction
It is widely known that hepatitis B virus (HBV) and hepatitis C virus (HCV) are among the most common blood-borne pathogens worldwide. Both viruses can lead to chronic infection leading to complications associated with liver-related morbidity and mortality. Chronic HBV infection is the leading cause of hepatocellular carcinoma (HCC) worldwide, while chronic HCV is the number one cause in the United States (US)[1],
[2]
. Guidelines for the management of chronic HBV are well established and recently updated, and treatment for chronic HCV infection has undergone a major update in recent years owing to the rapid development of direct-acting antivirals (DAAs). In addition, different modalities of outreach and care delivery of these new drugs that involve local pharmacies are currently being investigated to broaden the role of the pharmacist and perhaps increase cure rates.
Sources and selection criteria
To identify scientific studies for this review, the authors searched PubMed/MEDLINE, the Cochrane Library Database and clinical trial registries (clinicalTrials.gov) using the combination of search terms ‘chronic hepatitis B, chronic hepatitis C, cccDNA, functional and complete cure, nucleos(t)ide analogues, direct-acting antivirals, NS3/4A inhibitors, NS5A inhibitors, NS5B inhibitors’. Only English-language publications were selected. At least two reviewers independently evaluated each paper for relevance.
Hepatitis B epidemiology
It is estimated that more than 350 million individuals suffer from chronic HBV infection with one million deaths annually worldwide. In the United States, the overall prevalence is reported to be 700,000 to 2.2 million people (0.3–0.5%)[3],
[4],
[5]
. However, the true prevalence of chronic HBV infection in the US is likely to be several times higher because several patient populations (foreign-born, incarcerated and homeless individuals) are excluded from these estimates[1],
[6],
[7]
. While the implementation of HBV vaccine has led to a decline in the incidence of new HBV infection, the prevalence of chronic HBV infection still remains high.
Hepatitis B treatment updates
HBV is a member of the Hepadnaviridae family. It consists of a partially double-stranded relaxed circular DNA (rcDNA) genome. Upon entry into human hepatocytes, rcDNA is subsequently converted to a tightly coiled plasmid-like covalently closed circular DNA (cccDNA) in the host nucleus. cccDNA exists as an episomal minichromosome, and serves as the template for the transcription of viral RNAs and the production of virions. cccDNA is unusually stable and is able to avoid DNA-sensing cellular machinery. The production of tolerogenic proteins (HBsAg and hepatitis B e-antigen [HBeAg]) can lead to T-cell exhaustion. Hepatocytes have a long half-life, which allows for the maintenance of chronicity. Patients who recover from HBV infection and achieve HBsAg loss with or without seroconversion to HBsAb will harbour cccDNA in hepatocyte nuclei for life. This can potentially act as a reservoir for reactivation of viral genome replication when these patients undergo chemotherapy or other immunosuppressive therapies[8],
[9],
[10]
.
Sustained suppression of HBV improves clinical outcomes by slowing or reversing the degree of fibrosis and decreasing the incidence of HCC. Functional cure of chronic HBV is similar to resolved acute infection. There is HBsAg loss with or without HBsAb seroconversion with undetectable serum HBV DNA after stopping antiviral therapy. In functional cure, cccDNA persists, but it is not transcriptionally active. Current nucleos(t)ide therapy for HBV can lead to functional cure of 1–3% per year[8],
[11]
. While it can prevent the formation of new cccDNA, current therapy has no effect on existing cccDNA, and complete cure of HBV requires the elimination or permanent silencing of cccDNA in addition to functional cure[11]
. A better understanding of the virus’s lifecycle has led to the development of novel targets for the treatment of HBV with the goal of complete cure, Figure 1 illustrates the HBV lifecycle and potential therapeutic targets.
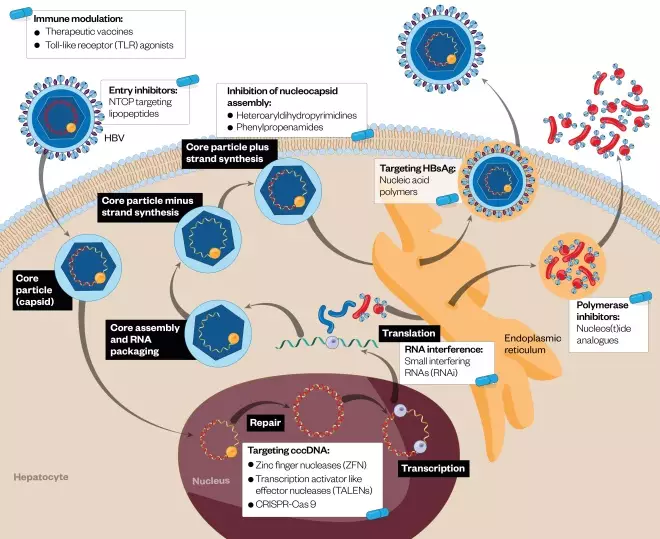
Figure 1: Hepatitis B virus lifecycle and potential therapeutic targets
Source: The Pharmaceutical Journal
Illustration of potential targets within the HBV lifecycle by novel antiviral agents. These include entry inhibitors, nucleocapsid and viral assembly inhibitors, covalently closed circular DNA (cccDNA) degradation, preventing secretion of hepatitis B surface antigen (HBsAg), viral RNA interference, and restoring host adaptive and innate immunity. Current nucleos(t)ide therapy works by inhibiting the reverse transcriptase activity of HBV polymerase. Novel nucleos(t)ides are also currently in development.
Current therapy
At present, there are seven treatments that have been approved for use in chronic HBV infection: two formulations of interferon and five nucleos(t)ide agents[12]
. Interferon mainly has immunomodulatory effects and limited direct antiviral effects. The current nucleos(t)ide agents approved for use are lamivudine, telbivudine, entecavir (ETV), adefovir dipivoxil and tenofovir disoproxil fumarate (TDF). These nucleos(t)ide analogues (NA) work by inhibiting the reverse transcriptase activity of HBV polymerase, thereby suppressing viral replication. First-line agents ETV and TDF have a high barrier to resistance and lead to complete viral suppression in 90% of patients after long-term use. While this can bring an improvement in fibrosis and reduce the incidence of HCC, very few patients achieve HBsAg loss. These therapies also do not affect cccDNA, and thus cannot lead to a true cure of chronic HBV infection[13],
[14]
. Most guidelines recommend lifelong therapy, with goals of alanine aminotransferase (ALT), also known as alanine transaminase, normalisation and viral suppression, which is the main disadvantage of these agents. Therefore, developing treatment strategies that can lead to a complete cure could eliminate potential adverse events associated with the long-term use of these nucleos(t)ide agents.
Novel nucleos(t)ide analogues (NAs)
New NAs are under investigation given the safety concerns of lifelong therapy. Long-term use of TDF, for instance, has been linked to renal toxic effects as well as a reduction in bone mineral density with an increase in markers of bone turnover[15],
[16],
[17]
.
Tenofovir alafenamide (TAF) is a prodrug of tenofovir that was recently approved for the treatment of chronic HBV mono-infection. When compared with TDF, TAF has demonstrated higher intracellular tenofovir levels, lower plasma tenofovir levels, and higher levels of the pharmacologically active metabolite tenofovir diphosphate[18],
[19]
. Multiple trials have demonstrated that patients receiving TAF have significantly smaller reductions in hip and spine bone mineral density at 48 weeks when compared with patients receiving TDF. In addition, there were minimal effects on markers of bone turnover as well as a significantly smaller reduction in glomerular filtration rate in these patients[15],
[16]
. A recent study in non-cirrhotic chronic HBV patients demonstrated that the change in serum HBV DNA levels in patients receiving TAF was similar to the change in patients receiving TDF[20]
.
Besifovir is a novel nucleotide phosphonate. In a recent trial comparing besifovir to ETV, there were no differences observed in the proportion of patients that achieved undetectable HBV DNA, ALT normalisation and HBeAg seroconversion[21]
. Both besifovir and TAF have an improved safety profile when compared with current NAs; however, like their predecessors, neither of these drugs affects cccDNA and cannot lead to cure.
Entry inhibitors
Viral entry into hepatocytes is mediated through the cellular receptor sodium taurocholate co-transporting polypeptide (NTCP). Myrcludex B is a novel synthetic lipopeptide that targets NTCP and blocks entry of HBV into the cell[22]
. In a phase IIa study, 75% of patients receiving myrcludex B for 12 weeks had a greater than 1 log decrease in HBV DNA, and 55% had ALT normalisation. The drug also showed excellent tolerability with few adverse events[23]
. Entry inhibitors may be particularly useful in the liver transplant setting to prevent infection of the new liver[14]
.
Capsid/viral assembly inhibitors
Assembly inhibitors work by destabilising the nucleocapsid and blocking RNA packaging, which produces empty capsids devoid of genetic information. Heteroaryldihydropyrimidines and phenylpropenamides are novel therapies that disrupt this assembly step in the HBV lifecycle[8],
[10]
. Heteroaryldihydropyrimidines inhibit HBV virion encapsidation by inhibiting capsid formation. For example, BAY 41-4109 is a heteroaryldihydropyrimidine that inhibits capsid formation, reduces the half-life of the core protein, and decreases the stability of normal capsids by misdirecting viral assembly[24]
. Phenylpropenamides induce changes in tertiary and quaternary structures of HBV capsid proteins that prevent encapsidation of viral pregenomic RNA into the nucleocapsid. AT-130 is a phenylpropenamide that decreases viral production by causing a premature initiation of virion assembly, which leads to normal capsids that are empty and non-infectious[25]
. It is unknown whether these agents affect HBsAg levels and further study is needed.
Targeting cccDNA
By degrading, silencing or eliminating cccDNA, therapies can block the replication and formation of new cccDNA. In an in vitro study in duck hepatocytes, zinc finger nucleases (ZFN) have been shown to block transcription of cccDNA by inhibiting rcDNA conversion to cccDNA and by directly destroying cccDNA[26]
. Transcription activator-like effector nucleases (TALENs) have the ability to cleave sequence-specific DNA targets[10],
[13]
. In a mouse model, TALENs decreased the production of HBeAg, HBsAg and hepatitis B core antigen [HBcAg], and suppressed the cccDNA level[27]
. The clustered regularly interspaced short palindromic repeats (CRISPR) associated Cas9 (CRISPR-Cas 9) system is a novel therapy that uses synthetic target RNA to direct site-specific cleavage of the cccDNA. This has been shown to reduce the production of HBV core and surface proteins[13],
[28]
.
Preventing HBsAg secretion
It is thought that during the early stages of HBV infection there are high circulating levels of HBsAg that can directly act on host dendritic cells, limiting cytokine production. This in turn causes T-cell exhaustion, leading to an immunosuppressed state. Decreasing the production of HBsAg may lead to a restoration of HBV-specific immune function[8]
. Inhibitors of HBsAg secretion have recently been identified and are currently in early phases of clinical trials. In a recent proof of concept study, nucleic acid polymers lowered HBsAg levels along with serum HBV DNA levels in chronic HBV-infected patients; however, these patients experienced viral rebound[29]
.
Targeting viral mRNA
Small interfering RNAs (siRNAs) within the RNA interference (RNAi) pathway induce gene silencing post-transcriptionally by binding to and inactivating viral mRNA, thereby preventing protein translation. In a mouse model, RNAi reduced the expression of HBsAg and HBeAg[30]
. In a phase IIa trial, chronic HBV patients who were taking ETV received one dose of ARC-520, a stable pooled siRNA molecule, which resulted in a 22% reduction of HBsAg levels when compared with the patients’ baseline[31]
. While there are concerns of drug toxicity with this molecule, this study still provided a proof of concept for it as a novel treatment method. If HBsAg levels are reduced, this in turn may lead to the resurrection of HBV-specific immune function.
Immune modulation
There are several different methods being investigated with the hope of restoring host adaptive and innate immunity to induce HBV clearance. Therapeutic vaccines that activate HBV-specific immune responses are currently being explored. Vaccines based on HBV proteins, HBV envelope subviral particles, HBsAg and HBcAg, dendritic cells (tarmogens), T-cell peptide epitopes and finally DNA are all undergoing clinical trials[10]
. Toll-like receptors (TLR) are important regulators of the innate and adaptive immune response, and it is known that HBV downregulates them[13],
[14]
. TLR agonists have therefore been studied as a method to enhance the host immune response. The TLR7 agonist GS 9620 induced long-term suppression of HBV DNA in a chimpanzee model and, in addition, serum HBsAg and HBeAg levels were decreased[32]
.
Hepatitis C epidemiology
Hepatitis C virus is the leading cause of liver-related morbidity and mortality, including end-stage liver disease and HCC, in the US. It is estimated that more than 150 million individuals are chronically infected with HCV worldwide[33]
. In the US, the overall prevalence is reported to be 2.7 to 3.9 million people (1.0–1.5%)[7],
[34],
[35]
. Similar to chronic HBV infection, the true disease burden of chronic HCV is likely to be several times higher because many populations with a high risk for HCV are excluded from these estimates. The prevalence in Western European countries, as well as Australia, is 1.25%; similar to those in the US[34]
. However, the global burden of HCV varies widely from country to country: a recent study demonstrated that the prevalence in Egypt was as high as 15%[36]
. More than 350,000 deaths are attributed to HCV-related end-stage liver disease and HCC annually[37],
[38]
.
Hepatitis C treatment updates
Development of direct-acting antivirals
The viral agent of hepatitis C, previously known as ‘non-A, non-B’ hepatitis, was identified as an RNA virus in 1989[7]
. Subsequently, HCV was classified into six major genotypes (1–6) and further subtypes that are closely related variants within the genotype. Genotype 1 is predominantly found in North American and European populations. Genotype 3 is the second most common genotype and accounts for the majority of infections in Asia, while North African and Middle Eastern populations are typically infected with genotype 4 virus[34]
.
Interferon therapy, which was approved by the Food and Drug Administration (FDA) in 1991, was the previous mainstay of therapy for more than 20 years. As a monotherapy it achieved sustained response rates of 20% after 48 weeks of treatment. Sustained virologic response (SVR) was assessed 24 weeks (SVR24) after the treatment course and considered achieved when the HCV RNA was below the lower limit of quantification. The combination of interferon with ribavirin improved SVR rates by up to 40%, with greater success in genotype 2 and 3 patients[39]
.
Development of pegylated interferon allowed for a single weekly injection and its co-administration with ribavirin further increased SVR rates to as high as 80% in genotype 3 patients, but remained under 50% for genotype 1 patients[40]
. Host genetic factors also contributed to the response to interferon therapy as patients with the CC genotype of the IL28B gene had an almost two-fold greater rate of SVR compared with patients with the TT genotype[41]
. Interferon therapy required weekly injections and patients suffered many side effects, such as flu-like symptoms, fever, myalgias, depression and cytopenia[41]
. Coupled with anaemia and fatigue from ribavirin therapy, completing the 48-week treatment course was challenging for many patients[42]
.
When the HCV encounters the host cell, viral particles are enveloped via their interaction with specific cell surface receptors. The viral and cellular membranes fuse, resulting in the release of the single-stranded RNA genome into the cytoplasm of the host cell. The viral genome encodes a large polyprotein that is cleaved by both host and viral proteases to produce ten viral proteins. This includes structural proteins (core, E1 and E1 glycoproteins), integral membrane protein (p7) and non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A, NS5B)[43]
. These non-structural proteins are essential for the intracellular processes of the virus lifecycle governing the replication, assembly and export of new virus particles. Inhibition of the non-structural (NS) proteins has become the major target in the development of direct-acting antiviral (DAA) agents. Figure 2 illustrates the HCV lifecycle and potential therapeutic targets.
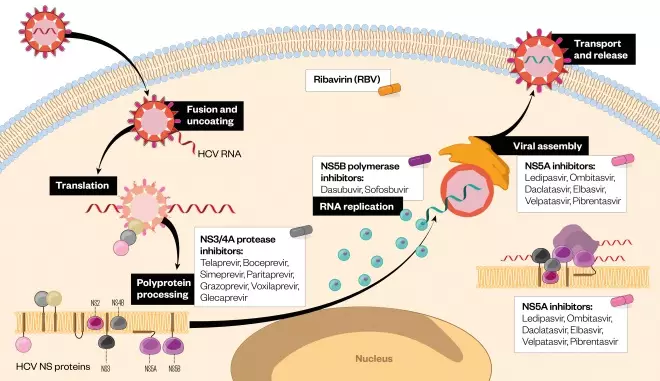
Figure 2: Hepatitis C virus lifecycle and potential therapeutic targets
Source: The Pharmaceutical Journal
Illustration of direct-acting antiviral (DAA) targets in the HCV lifecycle. These include NS3/4A protease inhibitors, which prevent polyprotein processing, NS5B inhibitors responsible for RNA-dependent RNA polymerase activity, and NS5A inhibitors that prevent viral assembly and formation of the RNA replication complex.
Together, the NS3/4A serine protease complex is responsible for cleaving the remaining NS proteins from the polyprotein complex. Inhibition of the serine protease activity prevents further processing and action of the remaining NS proteins[43]
. Boceprevir and telaprevir were the first generation of protease inhibitors released in 2011. Their use, in combination with interferon and ribavirin, resulted in higher SVR rates of upwards of 60% in previously untreated and treated patients[44],
[45]
. Unfortunately, these medications were poorly tolerated by patients and had low barriers to genetic resistance. Second-generation protease inhibitors include paritaprevir (administered in combination with dasabuvir and ombitasvir), simeprevir, grazoprevir (administered with elbasvir), voxilaprevir (administered with sofosbuvir and velpatasvir) and glecaprevir (administered with pibrentasvir) see Figure 2.
NS5B is an RNA-dependent RNA polymerase responsible for replicating the viral RNA genome. Sofosbuvir is a nucleoside analogue that works by binding to the enzyme active site and inducing a chain termination. As the NS5B protein is a highly conserved part of the HCV-genome, sofosbuvir is effective across all genotypes[46]
. Dasabuvir also functions to inhibit the NS5B protein but as a non-nucleoside inhibitor, it binds to one of four allosteric sites on the protein. Because the virus can more easily accommodate mutations at its allosteric sites, dasabuvir has a lower barrier to resistance than sofosbuvir and has a genotype specific to genotypes 1a and 1b[47]
.
NS5A is a membrane-associated phosphoprotein that is responsible for virion production, cell signalling and interferon response. It does not have any enzymatic activity but is believed to function via its interaction with other host cell factors and NS proteins including NS5B[48]
. NS5A protein has conserved regions (Sh3 binding domain) across all genotypes allowing for broad inhibitor activity[49]
. There are currently four NS5a inhibitors approved for use in the US: dalcatasvir, velpatasvir, ombitasvir and elbasvir (see Figure 2).
The FDA approval of DAAs has drastically changed the approach to the treatment of HCV. The benefits of DAAs include daily dosing with an oral medication, a limited side-effect profile, a shorter treatment course than interferon therapy and cure rates exceeding 90% in many populations. The most recent guidelines for the treatment of HCV set forth by the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA) recommend DAAs as the primary treatment for all genotypes of HCV[50]
. The key to success with antiviral therapy is the combination of agents that target more than one aspect of the HCV replication cycle and the use of drugs such as sofosbuvir that have a high genetic barrier to resistance. Genotype testing, history of prior HCV treatment (naïve versus experienced with interferon regimens), cirrhosis status (compensated versus decompensated), presence of chronic kidney disease (CKD) and HIV co-infection should all be taken into consideration when deciding on appropriate therapy.
Simeprevir/sofosbuvir
Simeprevir, used in combination with sofosbuvir (SMV/SOF), was one of the earliest DAA-based regimens indicated for the treatment of genotype 1 patients. It was successful for both treatment-naïve and treatment-experienced patients without cirrhosis yielding SVR rates of >90%[51],
[52]
. For patients with compensated cirrhosis, treatment for 24 weeks with or without ribavirin was recommended as an alternative therapy[53]
. With the advent of newer and more effective remedies, simeprevir is no longer prescribed in the UK and only rarely in the US.
Daclatasvir/sofosbuvir
In the US, daclatasvir/sofosbuvir (DCV/SOF) is recommended in treatment for genotype 3 patients without cirrhosis for 12 weeks, with an extension to 24 weeks in patients with cirrhosis. Guidelines are based on the phase III ALLY-3 trial that evaluated 12 weeks of DCV/SOF in genotype 3 patients who were either treatment-naïve or treatment-experienced. This included patients with compensated cirrhosis. Overall, SVR12 rates were 89%, although it was less than 70% for patients with cirrhosis[54]
.
In the subsequent ALLY-3+ trial there was no significant difference noted between 12 weeks (88%) and 16 weeks (92%) of treatment with DCV/SOF plus ribavirin in patients with compensated cirrhosis[55]
. The French Multicenter Compassionate use trial is a real-world study that compared 12 versus 24 weeks of DCV/SOF with or without ribavirin in cirrhotic patients. The majority of patients received 24 weeks of therapy. Overall, SVR12 rates in the 24-week treatment group were 89%; 98% in those without cirrhosis and 86% in those with cirrhosis. The addition of ribavirin provided no statistically significant benefit[56]
.
DCV/SOF is not commonly used for treatment of genotype 1 patients but can be considered for treatment-naïve or treatment-experienced patients without cirrhosis. In the UK it is recommended for genotype 1 patients without cirrhosis but who have significant fibrosis[57]
. The combination of DCV/SOF for treatment of genotype 1 patients has been evaluated as part of an open-label phase II study conducted with the AI444040 Study Group. The overall SVR12 rate for treatment-naïve patients was 98%. In the cohort of treatment-experienced patients, all received 24 weeks of treatment and 97.5% achieved SVR12. Based on these results, a 12-week course of DCV/SOF without ribavirin is sufficient for patients without cirrhosis regardless of prior treatment status[58]
.
Ledipasvir/sofosbuvir
Ledipasvir, as a fixed-dose pill in combination with sofosbuvir (LDV/SOF), is approved for treatment for genotypes 1, 4, 5 and 6. In the ION-1 trial, 97% of treatment-naïve genotype 1 patients achieved SVR12 whether treated for 12 weeks or 24 weeks with once-daily LDV/SOF. Favourable outcomes were also seen in patients with compensated cirrhosis achieving SVR12 rates >94%[59]
. The subsequent ION-3 trial investigated shortening the treatment course to 8 weeks in treatment-naïve genotype 1 patients without cirrhosis. No significant difference was found in SVR12 rates whether treating for 12 weeks (95%) or 8 weeks (94%)[60]
. Based on this study, 8 weeks of LDV/SOF has been approved in the US for treatment naïve, non-cirrhotic patients with viral load < 6M IU/ml. In the UK, 8 weeks of LDV/SOF is the preferred therapy for treatment-naïve genotype 1 patients without cirrhosis regardless of viral load[61]
.
Treatment-experienced patients without cirrhosis can also be effectively treated for a 12-week course of LDV/SOF as demonstrated in the ION-2 trial with overall SVR12 rates of 94%[62]
. Further data come from the SIRIUS trial, which specifically evaluated LDV/SOF in treatment-experienced patients with cirrhosis. Those receiving LDV/SOF plus weight-based ribavirin for 12 weeks achieved SVR12 rates (96%) equal to those treated with LDV/SOF for 24 weeks (97%)[63]
. Treatment-experienced patients with compensated cirrhosis can be effectively treated with LDV/SOF plus ribavirin for 12 weeks or 24 weeks without ribavirin as an alternative. In addition to genotype 1, several studies evaluated LDV/SOF in genotype 4 and 5 patients and demonstrated similar high SVR12 rates of 95%[64],
[65]
.
Retrospective analysis of real-world data supports the effectiveness of LDV/SOF in curing hepatitis in patients treated outside of clinical trials. The data from the TRIO health network, which consists of more than 1,500 patients, demonstrated SVR12 rates of 94%[66]
. The HCV-TARGET study evaluated the use of LDV/SOF for 8, 12 and 24 weeks in another real-world cohort. There were no significant differences in SVR12 rates among the eligible treatment groups (8 weeks 96%, 12 weeks 97%, 24 weeks 95%)[67]
. Both these studies demonstrate that real-world use of DAAs is equivalent to clinical trial results.
Elbasvir/grazoprevir
Elbasvir/grazoprevir (EBR/GZR) is a fixed-dose combination NS5A inhibitor (EBR) and NS3/4a protease inhibitor (GZR) approved for the treatment of genotype 1 and 4 patients, including those with compensated cirrhosis. The series of phase II C-WORTHY trials demonstrated the safety and efficacy of a 12-week course of EBR/GZR for treatment-naïve and previously interferon/ribavirin-experienced genotype 1 patients with SVR12 rates of 93–98% across all treatment groups[68]
.
An important take away from these trials was that an attempt to treat this population for an 8-week course resulted in an SVR12 rate of only 80%, with 17% virologic failure, indicating that a shortened treatment course was not adequate[69],. The results of the phase III C-EDGE trial further supported these findings, with an overall SVR rate of 92% and 99% for treatment-naïve genotype 1a and 1b patients, respectively. Among genotype 1a patients, the presence of resistance-associated variants (RAVs) at positions M28, Q30, L31 or Y93 was found to be associated with a reduced response to treatment[70]
.
Subsequent trials in the C-WORTHY series as well as the phase III C-EDGE-TE trial corroborated the findings of reduced SVR12 rates in genotype 1a patients with RAVs. However, equivalent SVR12 rates were seen in this patient population when the therapy was extended and ribavirin was added to their regimen[71],
[72]
. As a result, resistance testing is recommended for all genotype 1a patients prior to initiation of therapy with EBR/GZR. If baseline polymorphisms are detected at positions 28, 30, 31 or 93, the indicated treatment is 16 weeks, with the addition of ribavirin. Genotype 1a patients without resistance mutations and all genotype 1b regardless of cirrhosis status can be effectively treated for 12 weeks.
Paritaprevir/ritonavir/ombitasvir/dasabuvir
Paritaprevir is an NS3/4a inhibitor that when administered with ritonavir, a CYP3A4 inhibitor, attains increased plasma levels and a prolonged half-life allowing for once-daily dosing. Both the NS5A inhibitor ombitasvir and dasabuvir, the non-nucleoside NS5B inhibitor, have potent in vitro activity against genotype 1 virus. The fixed-dose combination pill of paritaprevir/ritonavir/ombitasvir plus dasabuvir (PrOD) is available for the treatment of genotype 1 patients, with a better effect profile in treating genotype 1b patients.
The PEARL III and IV trials evaluated 12 weeks of therapy with or without ribavirin in treatment-naïve genotype 1 patients without compensated cirrhosis. For genotype 1a patients, the addition of ribavirin to a 12-week course increased SVR12 rates to 97% compared with 90% when ribavirin was not administered. Genotype 1b patients had SVR rates of greater than 99% regardless of whether they had received ribavirin or not[73]
. TURQUOISE II enrolled both treatment-naïve and treatment-experienced patients with compensated cirrhosis for a 12- or 24-week treatment course of PrOD plus ribavirin. The main statistically significant difference in outcome was noted for genotype 1a patients. Those treated for 12 weeks had an SVR rate of 88.6% compared with 94.2% when treated for 24 weeks.
Based on these findings, the AASLD recommends treating genotype 1a patients with compensated cirrhosis for 24 weeks of PrOD plus ribavirin. Similarly, in non-cirrhotic patients, ribavirin is added to the 12-week course for genotype 1a patients given the results of the PEARL trials. All genotype 1b patients regardless of cirrhosis status or treatment history can be effectively treated with a 12-week course of PrOD[74]
, which was recently approved for once-daily dosing.
Despite only having a global prevalence of 13%, genotype 4 is the most common genotype in North Africa and the Middle East. Along with EBR/GZR and LDV/SOF, the combination of paritaprevir/ritonavir/ombitasvir plus ribavirin is also recommended for treatment of all genotype 4 patients. This recommendation comes from results of the PEARL-1 trial, which enrolled both treatment-naïve and treatment-experienced genotype 4 patients for 12 weeks of paritaprevir/ritonavir/ombitasvir with or without ribavirin, with SVR12 rates of 100%[75]
. A 12-week treatment course with ribavirin is also recommended for genotype 4 patients with compensated cirrhosis. Results of the ongoing AGATE trials reveal high SVR rates of 96% and 100% in treatment-experienced or treatment-naïve cirrhotic patients treated for 12 or 16 weeks, respectively[76]
.
Sofosbuvir/velpatasvir
The fixed-dose combination of sofosbuvir/velpatasvir (SOF/VEL) is the first DAA to receive FDA approval in treatment of all genotypes and is first-line therapy for genotype 3 patients. Genotype 3 has traditionally been more challenging to treat with higher relapse rates and patients exhibiting higher risk of progression to cirrhosis and HCC compared with other genotypes. In the series of ASTRAL trials, treatment with SOF/VEL for 12 weeks resulted in SVR12 rates of >95% across all genotypes. The ASTRAL-1 trial enrolled genotype 1, 2, 4, 5 and 6 patients, both treatment-naïve and treatment-experienced, but excluded those treated with an NS5B or NS5A inhibitor. The overall SVR12 rate was 99%[77]
.
Two comparison trials (ASTRAL-2 and ASTRAL-3) were also conducted with genotype 2 and 3 patients, comparing SOF/VEL to SOF plus ribavirin (standard care at the time). For genotype 2 patients, the SVR12 rate was 99% for those treated with SOF/VEL compared with 94% after 12 weeks of SOF plus ribavirin. In the genotype 3 cohort, 95% of patients treated with SOF/VEL achieved SVR12, a significant increase over the 80% of patients treated with SOF plus ribavirin for 24 weeks.
Even in the traditionally hard-to-treat cohorts, 90% of patients with compensated cirrhosis achieved SVR12, as did 90% of those previously treated on interferon regimens. The results of the ALLY-3 trials in treating genotype 3 patients with compensated cirrhosis demonstrated the benefit of adding ribavirin to the treatment regimen to improve SVR12 rates. Given the difficulty and imperative in treating this group of patients, American Association for the Study of Liver Diseases (AASLD) guidelines recommend adding ribavirin to SOF/VEL for compensated cirrhotic patients with genotype 3[78]
.
Treatment of patients with decompensated cirrhosis
Before the advent of DAAs, treating HCV in patients with advanced cirrhosis or a prior liver transplant posed a difficult challenge. Patients with decompensated cirrhosis had significantly lower cure rates and interferon therapy was contraindicated for those on chronic immunosuppression. The use of protease inhibitors is also contraindicated for treatment in patients with decompensated cirrhosis due to the risk of increased liver enzymes, hyperbilirubinemia and worsening hepatic disease. The pan-genotypic NS5B inhibitor sofosbuvir, in combination with NS5A inhibitors, has become the mainstay of treatment for HCV patients with advanced liver disease, with cure rates approaching those of patients without cirrhosis.
LDV/SOF with ribavirin for 12 weeks is recommended to treat genotype 1 and 4 patients with decompensated cirrhosis or a history of liver transplant. The results of the open-label phase II SOLAR-1 study demonstrated SVR rates of greater than 85% for genotype 1 patients who received either 12 or 24 weeks of LDV/SOF plus ribavirin. Of note, those who achieved SVR also had improvements in their Child-Turcotte-Pugh (CTP) and Model for End-Stage Liver Disease (MELD) scores. The study also enrolled a separate cohort of patients who had previously undergone liver transplant, stratified according to cirrhosis status. Those without cirrhosis had SVR12 rates greater than 95% regardless of treatment duration[79]
.
The SOLAR-2 study expanded upon the results of SOLAR-1, again enrolling those with advanced cirrhosis or who were post-liver transplant, but this time including more patients with genotype 4. Similarly, SVR12 rates of 85–88% were seen in CTP class B and C patients treated for 12 or 24 weeks with LDV/SOF plus ribavirin. Current AASLD guidelines recommend 12 weeks of LDV/SOF plus ribavirin in this population but, if tolerated, practitioners may want to extend the treatment course to 24 weeks for increased benefit[80]
.
The recent development of a pan-genotypic regimen of sofosbuvir combined with a next-generation NS5A inhibitor velpatasvir (SOF/VEL) has expanded the treatment options for patients with decompensated cirrhosis regardless of genotype. The ASTRAL-4 trial enrolled genotype 1–6 patients with CPT B cirrhosis and no history of liver transplant. Overall, patients treated with SOF/VEL plus ribavirin for 12 weeks achieved SVR12 rates of 94%. In particular, those patients with genotype 3 infection had SVR12 rates of 85% with this treatment regimen. All patients with genotypes 2, 4 and 6 achieved SVR12 except for one patient in the 24-week treatment arm who died of liver failure after completing 28 days of treatment. Consistent with these findings, the AASLD guidelines recommend SOF/VEL plus ribavirin for 12 weeks in treatment of genotype 1–4 patients with decompensated cirrhosis[81]
.
Treatment of patients with renal disease
The prevalence of HCV is higher in patients on long-term haemodialysis. This is due to a series of risk factors including number of blood transfusions (before effective testing for HCV), time on dialysis and history of renal transplant[82]
. Conversely, patients with HCV infection are at increased risk for the development of CKD due to extra-hepatic manifestations including cryoglobulinemia, diabetes and membranoproliferative glomerulonephritis (MPGN).
A meta-analysis of haemodialysis-dependent patients found that not only does HCV increase the incidence of liver-related deaths but patients with HCV also have increased risk of death from cardiovascular disease[83]
. For patients with CKD that ultimately undergo renal transplantation, concomitant HCV infection is associated with increased mortality and reduced graft survival compared with transplanted patients without HCV[84]
.
These findings highlight the importance of effective HCV treatment for this population. Due to significantly adverse events and the toxicity of interferon therapy in this patient population, many patients with CKD previously went untreated. While all DAAs can be administered to patients with mild renal impairment, only EBR/GZR and PrOD have been proven safe and effective in patients with severe renal impairment defined as creatinine clearance ≤30, end-stage renal disease on haemodialysis, or a history of renal transplant.
The C-SURFER trial demonstrated the safety and efficacy of once-daily EBR/GZR for 12 weeks in genotype 1 patients with CKD (stage 4–5), 76% of which were on haemodialysis. SVR12 rates were 99% and the medication was well tolerated, with the most common adverse events being headache, nausea and vomiting[85]
. PrOD is also available for treatment of genotype 1 patients with CKD and these medications undergo hepatic metabolism with minimal renal clearance. In the RUBY-1 trial, patients with stage 4–5 CKD were treated with PrOD for 12 weeks with or without ribavirin. Overall SVR12 rates were 90% in this patient population[86]
. Based on these studies, EBR/GZR and PrOD are the only recommended regimens for patients with chronic HCV infection with impaired renal function, including those patients on haemodialysis.
Treatment of patients with HIV/HCV co-infection
The importance of treating chronic HCV in HIV/HCV co-infected patients has been demonstrated in successful cures with PEG-interferon/ribavirin that resulted in an improvement in fibrosis along with lower rates of hepatic decompensation, HCC, and liver-related morbidity and mortality[87],
[88],
[89]
. SVR12 was significantly harder to achieve in this patient population when compared with mono-infected individuals in the interferon era. However, with the advent of DAAs, SVR12 rates now achieve >95% and are equivalent to mono-infected patients[90],
[91]
. Treatment regimens for patients with HIV/HCV co-infection are no different than regimens used for HCV mono-infected patients with the same genotype. DCV/SOF, LDV/SOF and SOF/VEL have all been shown in clinical trials to have SVR12 rates of ≥95%[90],
[92],
[93]
.
Real-world data have also demonstrated the safety and efficacy of these regimens in HIV/HCV co-infected individuals. An analysis from the TRIO network reported an overall SVR12 rate of 98% in patients with HIV/HCV co-infection treated with LDV/SOF for either 8, 12 or 24 weeks[94]
. The GECCO-01 real-world cohort further evaluated LDV/SOF for 8 weeks in HIV/HCV co-infected and HCV mono-infected patients who were treatment-naïve, non-cirrhotic, with HCV genotype 1 and low viral load (<6 million IU/mL). SVR12 rates were equivalent among the two groups: co-infected 96%, mono-infected 99%[95]
. However, despite these recent results, 8 weeks of therapy for HIV/HCV co-infected patients is not currently recommended. In selecting appropriate therapy, providers need to be aware of potential drug–drug interactions between anti-retrovirals and DAAs. It is not recommended that HIV therapy be interrupted to allow for treatment of hepatitis C.
The next frontier of DAA therapy
The use of DAAs has allowed for effective and well-tolerated treatment options for patients with HCV. The next focus of DAA therapy is the ongoing development of pan-genotypic regimens, treatment of patients who failed other DAA-based therapies and the feasibility of shortened treatment courses. Although many DAAs have resulted in cure rates exceeding 90% in many populations, effectively treating the 10% or so of patients that failed therapy has presented a challenge. Many of these patients develop RAVs after exposure to NS5As or NS3/4a inhibitors, which may limit their further treatment options.
Voxilaprevir (VOX) is a next-generation NS3/4A protease inhibitor that is used in combination with sofosbuvir plus velpatasvir (SOF/VEL/VOX) and has activity against genotypes 1–6. In an open-label phase II study, the efficacy of this regimen was evaluated in genotype 1 treatment-naïve and DAA-experienced patients. They were treated for 6–12 weeks with SOF/VEL/VOX. In treatment-naïve patients, those without cirrhosis had an SVR12 rate of 71% after 6 weeks of treatment compared with 100% when treated for 8 weeks. For treatment-naïve patients with compensated cirrhosis, 94% achieved SVR12. The addition of ribavirin to this group showed no improvement in SVR12 rates. All DAA-experienced patients achieved SVR12 regardless of cirrhosis status when treated for 12 weeks. Baseline RAVs were detected in 82% of those previously treated with DAAs, and for those with prior exposure to NS5A, 93% had RAVs[96]
. While there are still more studies investigating this regimen in development, these results suggest that 8 weeks may prove an effective regimen for certain patient populations such as treatment-naïve patients, including those with compensated cirrhosis. For treatment-experienced patients, 12 weeks appears to be the optimal treatment length. Furthermore, the combination of three pan-genotypic agents results in a therapy with a high genetic barrier to resistance that may be effective in treating patients who failed prior therapies.
The combination of the next-generation NS3/4A protease inhibitor glecaprevir (formerly known as ABT-493) and the next-generation NS5A inhibitor pibrentasvir (formerly known as ABT-530) has demonstrated activity against all genotypes and against frequently observed variants that have been resistant to other NS5A inhibitors[97]
. In the SURVEYOR trials, 12 weeks of treatment with glecaprevir/pibrentasvir (G/P) in non-cirrhotic genotype 1 patients resulted in overall SVR12 rates >98%. Reducing treatment duration to 8 weeks was equally effective in this population[98]
. A subsequent trial evaluated its efficacy in genotype 1 and 3 patients with compensated cirrhosis. SVR12 rates were 96% in genotype 1 patients and 98% across all genotype 3 patients. The presence of baseline RAVs had no significant impact on SVR rates[99]
.
Conclusion
Chronic HBV and HCV remain a significant global burden affecting more than 400 million people worldwide. Better understanding of the lifecycle of these viruses has led to the development of novel therapies. In chronic HBV infection, it is likely that combination therapy will be needed to obtain a functional or complete cure. In chronic HCV infection, effective therapy now exists for all populations regardless of severity of liver disease, HIV co-infection or other co-morbidities such as CKD. As research into HCV treatment continues, we can expect shorter treatment courses and cure rates approaching 100% for all populations.
Author disclosures and conflicts of interest:
Douglas Dieterich has served as a consultant or scientific advisor for AbbVie, Bristol-Myers Squibb, Gilead Sciences Inc, Idenix Pharmaceuticals Inc, Merck & Co Inc, and Janssen Therapeutics. He has received grants or research support from AbbVie, Bristol-Myers Squibb, Gilead Sciences Inc, Merck & Co Inc, and Janssen Therapeutics. The remaining authors have no conflicts of interest. No writing assistance was used in the production of this manuscript.
Reading this article counts towards your CPD
You can use the following forms to record your learning and action points from this article from Pharmaceutical Journal Publications.
Your CPD module results are stored against your account here at The Pharmaceutical Journal. You must be registered and logged into the site to do this. To review your module results, go to the ‘My Account’ tab and then ‘My CPD’.
Any training, learning or development activities that you undertake for CPD can also be recorded as evidence as part of your RPS Faculty practice-based portfolio when preparing for Faculty membership. To start your RPS Faculty journey today, access the portfolio and tools at www.rpharms.com/Faculty
If your learning was planned in advance, please click:
If your learning was spontaneous, please click:
References
[1] Martin P, Lau DT-Y, Nguyen MH et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2015 update. Clin Gastroenterol Hepatol 2015;13(12):2071–2087.e16. doi: 10.1016/j.cgh.2015.07.007
[2] Moyer VA & U.S. Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2013;159(5):349–357. doi: 10.7326/0003-4819-159-5-201309030-00672
[3] Montuclard C, Hamza S, Rollot F et al. Causes of death in people with chronic HBV infection: A population-based cohort study. J Hepatol 2015;62(6):1265–1271. doi: 10.1016/j.jhep.2015.01.020
[4] Terrault NA, Bzowej NH, Chang K-M et al. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016;63(1):261–283. doi: 10.1002/hep.28156
[5] LeFevre ML & U.S. Preventive Services Task Force. Screening for hepatitis B virus infection in nonpregnant adolescents and adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2014;161(1):58–66. doi: 10.7326/M14-1018
[6] Gish RG, Cohen CA, Block JM et al. Data supporting updating estimates of the prevalence of chronic hepatitis B and C in the United States. Hepatology 2015;62(5):1339–1341. doi: 10.1002/hep.28026
[7] Sarpel D, Baichoo E & Dieterich DT. Chronic hepatitis B and C infection in the United States: a review of current guidelines, disease burden and cost effectiveness of screening. Expert Rev Anti Infect Ther 2016;14(5):511–521. doi: 10.1586/14787210.2016.1174066
[8] Wilson EMP, Tang L & Kottilil S. Eradication strategies for chronic hepatitis B infection. Clin Infect Dis 2016;62 Suppl 4: S318–325. doi: 10.1093/cid/ciw044
[9] Lucifora J & Protzer U. Attacking hepatitis B virus cccDNA–The holy grail to hepatitis B cure. J Hepatol 2016;64(1 Suppl):S41–48. doi: 10.1016/j.jhep.2016.02.009
[10] Ward H, Tang L, Poonia B et al. Treatment of hepatitis B virus: an update. Future Microbiol 2016;11(12):1581–1597. doi: 10.2217/fmb-2016-0128
[11] Durantel D & Zoulim F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J Hepatol 2016;64(1):S117–131. doi: 10.1016/j.jhep.2016.02.016
[12] Lok ASF, McMahon BJ, Brown RS et al. Antiviral therapy for chronic hepatitis B viral infection in adults: a systematic review and meta-analysis. Hepatology 2016;63(1):284–306. doi: 10.1002/hep.28280
[13] Lin C-L, Yang H-C, Kao J-H. Hepatitis B virus: new therapeutic perspectives. Liver Int 2016;36:85–92. doi: 10.1111/liv.13003
[14] Brahmania M, Feld J, Arif A et al. New therapeutic agents for chronic hepatitis B. Lancet Infect Dis 2016;16(2):e10–21. doi: 10.1016/S1473-3099(15)00436-3
[15] Chan HLY, Fung S, Seto WK et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg-positive chronic hepatitis B virus infection: a randomised, double-blind, phase III, non-inferiority trial. Lancet Gastroenterol Hepatol 2016;1(3):185–195. doi: 10.1016/S2468-1253(16)30024-3
[16] Buti M, Gane E, Seto WK et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of patients with HBeAg-negative chronic hepatitis B virus infection: a randomised, double-blind, phase III, non-inferiority trial. Lancet Gastroenterol Hepatol 2016;1(3):196–206. doi: 10.1016/S2468-1253(16)30107-8
[17] Brouwer WP. Tenofovir alafenamide for hepatitis B: evolution or revolution? Lancet Gastroenterol Hepatol 2016;1(3):174–175. doi: 10.1016/S2468-1253(16)30083-8
[18] Markowitz M, Zolopa A, Squires K et al. Phase I/II study of the pharmacokinetics, safety and antiretroviral activity of tenofovir alafenamide, a new prodrug of the HIV reverse transcriptase inhibitor tenofovir, in HIV-infected adults. J Antimicrob Chemother 2014;69(5):1362–1369. doi: 10.1093/jac/dkt532
[19] Murakami E, Wang T, Park Y et al. Implications of efficient hepatic delivery by tenofovir alafenamide (GS-7340) for hepatitis B virus therapy. Antimicrob Agents Chemother 2015;59(6):3563–3569. doi: 10.1128/AAC.00128-15
[20] Agarwal K, Fung SK, Nguyen TT et al. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J Hepatol 2015;62(3):533–540. doi: 10.1016/j.jhep.2014.10.035
[21] Yuen M-F, Ahn SH, Lee KS et al. Two-year treatment outcome of chronic hepatitis B infection treated with besifovir vs. entecavir: results from a multicentre study. J Hepatol 2015;62(3):526–532. doi: 10.1016/j.jhep.2014.10.026
[22] Seto WK & Yuen MF. New pharmacological approaches to a functional cure of hepatitis B. Clin Liver Dis 2016;8(4):83–88. doi: 10.1002/cld.577
[23] Bogomolov P, Voronkova N, Allweiss L et al. A proof-of-concept phase IIa clinical trial with HBV/HDV entry inhibitor Myrcludex B. Hepatology 2014;60(6):1279A–1280A. doi: 10.1002/hep.27588
[24] Brezillon N, Brunelle MN, Massinet H et al. Antiviral activity of Bay 41-4109 on hepatitis B virus in humanized Alb-uPA/SCID mice. Albert ML, editor. PLoS One 2011;6(12):e25096. doi: 10.1371/journal.pone.0025096
[25] Katen SP, Tan Z, Chirapu SR et al. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure 2013;21(8):1406–1416. doi: 10.1016/j.str.2013.06.013
[26] Zimmerman KA, Fischer KP, Joyce MA et al. Zinc finger proteins designed to specifically target duck hepatitis B virus covalently closed circular DNA inhibit viral transcription in tissue culture. J Virol 2008;82(16):8013–8021. doi: 10.1128/JVI.00366-08
[27] Chen J, Zhang W, Lin J et al. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol Ther 2014;22(2):303–311. doi: 10.1038/mt.2013.212
[28] Lin SR, Yang HC, Kuo Y-T et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol Ther Nucleic Acids 2014;3:e186. doi: 10.1038/mtna.2014.38
[29] Al-Mahtab M, Bazinet M, Vaillant A. Effects of nucleic acid polymer therapy alone or in combination with immunotherapy on the establishment of SVR in patients with chronic HBV infection. J Clin Virol 2015;69:228. doi: 10.1016/j.jcv.2015.06.021
[30] McCaffrey AP, Nakai H, Pandey K et al. Inhibition of hepatitis B virus in mice by RNA interference. Nat Biotechnol 2003;21(6):639–644. doi: 10.1038/nbt824
[31] Gish RG, Yuen MF, Chan HLY et al. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antiviral Res 2015;121:97–108. doi: 10.1016/j.antiviral.2015.06.019
[32] Lanford RE, Guerra B, Chavez D et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013;144(7):1508–17,1517–10. doi: 10.1053/j.gastro.2013.02.003
[33] Shire NJ & Sherman KE. Epidemiology of hepatitis C virus: a battle on new frontiers. Gastroenterol Clin North Am 2015;44(4):699–716. doi: 10.1016/j.gtc.2015.07.002
[34] Gower E, Estes C, Blach S et al. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 2014;61(1):S45–57. doi: 10.1016/j.jhep.2014.07.027
[35] Smith BD, Morgan RL, Beckett GA et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm reports Morb Mortal Wkly report Recomm reports 2012;61(RR-4):1–32.
[36] Mohamoud YA, Mumtaz GR, Riome S, Miller DW & Abu-Raddad LJ. The epidemiology of hepatitis C virus in Egypt: a systematic review and data synthesis. BMC Infectious Diseases 2013;13:288. doi: 10.1186/1471-2334-13-288
[37] Mohd Hanafiah K, Groeger J, Flaxman AD et al. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013;57(4):1333–1342. doi: 10.1002/hep.26141
[38] Lozano R, Naghavi M, Foreman K et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2095–2128. doi: 10.1016/S0140-6736(12)61728-0
[39] Poynard T, Marcellin P, Lee SS et al. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT). Lancet 1998;352(9138):1426–1432. doi: 10.1016/S0140-6736(98)07124-4
[40] Manns MP, McHutchison JG, Gordon SC et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001;358(9286):958–965. doi: 10.1016/S0140-6736(01)06102-5
[41] Dusheiko G. Side effects of  α interferon in chronic hepatitis C. Hepatology 1997;26(S3):112S–121S. doi: 10.1002/hep.510260720
[42] Feld JJ & Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005;436(7053):967–972. doi: 10.1038/nature04082
[43] Lindenbach BD & Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature 2005;436(7053):933–938. doi: 10.1038/nature04077
[44] Poordad F, McCone J, Bacon BR et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011;364(13):1195–1206. doi: 10.1056/NEJMoa1010494
[45] Vierling JM, Davis M, Flamm S et al. Boceprevir for chronic HCV genotype 1 infection in patients with prior treatment failure to peginterferon/ribavirin, including prior null response. J Hepatol 2014;60(4):748–756. doi: 10.1016/j.jhep.2013.12.013
[46] Burton JR & Everson GT. HCV NS5B polymerase inhibitors. Clin Liver Dis 2009;13(3):453–465. doi: 10.1016/j.cld.2009.05.001
[47] Schmidt WN, Nelson DR, Pawlotsky JM et al. Direct-acting antiviral agents and the path to interferon independence. Clin Gastroenterol Hepatol 2014;12(5):728–737. doi: 10.1016/j.cgh.2013.06.024
[48] Lemm JA, O’Boyle D, Liu M et al. Identification of hepatitis C virus NS5A inhibitors. J Virol 2010;84(1):482–491. doi: 10.1128/JVI.01360-09
[49] Szabo G. Hepatitis C virus NS5A protein–a master regulator? Gastroenterology 2006;130(3):995–999. doi: 10.1053/j.gastro.2006.01.072
[50] AASLD/IDSA HCV Guidance Panel. Hepatitis C guidance: AASLD-IDSA recommendations for testing, managing, and treating adults infected with hepatitis C virus. Hepatology 2015;62(3):932–954. doi: 10.1002/hep.27950
[51] Kwo P, Gitlin N, Nahass R et al. Simeprevir plus sofosbuvir (12 and 8 weeks) in hepatitis C virus genotype 1-infected patients without cirrhosis: OPTIMIST-1, a phase III, randomized study. Hepatology 2016;64(2):370–380. doi:10.1002/hep.28467
[52] Lawitz E, Sulkowski MS, Ghalib R et al. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet 2014;384(9956):1756–1765. doi: 10.1016/S0140-6736(14)61036-9
[53] Lawitz E, Matusow G, DeJesus E et al. Simeprevir plus sofosbuvir in patients with chronic hepatitis C virus genotype 1 infection and cirrhosis: a phase III study (OPTIMIST-2). Hepatology 2016;64(2):360–369. doi: 10.1002/hep.28422
[54] Nelson DR, Cooper JN, Lalezari JP et al. All-oral 12-week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY-3 phase III study. Hepatology 2015;61(4):1127–1135. doi: 10.1002/hep.27726
[55] Leroy V, Angus P, Bronowicki J-P et al. Daclatasvir, sofosbuvir, and ribavirin for hepatitis C virus genotype 3 and advanced liver disease: a randomized phase III study (ALLY-3+). Hepatology 2016;63(5):1430–1441. doi: 10.1002/hep.28473
[56] Hézode C, Lebray P, De Ledinghen V et al. Daclatasvir plus sofosbuvir, with or without ribavirin, for hepatitis C virus genotype 3 in a French early access programme. Liver Int 2017; 1–11; doi: 10.1111/liv.13383
[57] National Institute for Health and Care Excellence. Daclatasvir for treating chronic hepatitis C. November 2015. Available at: https://www.nice.org.uk/guidance/ta364 (accessed August 2017).
[58] Sulkowski MS, Gardiner DF, Rodriguez-Torres M et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med 2014;370(3):211–221. doi: 10.1056/NEJMoa1306218
[59] Afdhal N, Zeuzem S, Kwo P et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014;370(20):1889–1898. doi:10.1056/NEJMoa1402454
[60] Kowdley KV, Gordon SC, Reddy KR et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med 2014;370(20):1879–1888. doi: 10.1056/NEJMoa1402454
[61] National Institute for Health and Care Excellence. Ledipasvir–sofosbuvir for treating chronic hepatitis C. November 2015. Available at: https://www.nice.org.uk/guidance/ta363 (accessed August 2017).
[62] Afdhal N, Reddy KR, Nelson DR et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 2014;370(16):1483–1493. doi: 10.1056/NEJMoa1316366
[63] Bourlière M, Bronowicki JP, de Ledinghen V et al. Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: a randomised, double-blind, phase II trial (SIRIUS). Lancet Infect Dis 2015;15(4):397–404. doi: 10.1016/S1473-3099(15)70050-2
[64] Kohli A, Kapoor R, Sims Z et al. Ledipasvir and sofosbuvir for hepatitis C genotype 4: a proof-of-concept, single-centre, open-label phase IIa cohort study. Lancet Infect Dis 2015;15(9):1049–1054. doi: 10.1016/S1473-3099(15)00157-7
[65] Abergel A, Asselah T, Metivier S et al. Ledipasvir-sofosbuvir in patients with hepatitis C virus genotype 5 infection: an open-label, multicentre, single-arm, phase II study. Lancet Infect Dis 2016;16(4):459–464. doi: 10.1016/S1473-3099(15)00529-0
[66] Tapper EB, Bacon BR, Curry MP et al. Real-world effectiveness for 12 weeks of ledipasvir-sofosbuvir for genotype 1 hepatitis C: the Trio Health study. J Viral Hepat 2017;24(1):22–27. doi: 10.1111/jvh.12611
[67] Terrault NA, Zeuzem S, Di Bisceglie AM et al. Effectiveness of ledipasvir-sofosbuvir combination in patients with hepatitis C virus infection and factors associated with sustained virologic response. Gastroenterology 2016;151(6):1131–1140.e5. doi: 10.1053/j.gastro.2016.08.004
[68] Sulkowski M, Hezode C, Gerstoft J, et al. Efficacy and safety of 8 weeks versus 12 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin in patients with hepatitis C virus genotype 1 mono-infection and HIV/hepatitis C virus co-infection (C-WORTHY): a randomised, open-label phase II trial. Lancet 2015;385(9973):1087–1097. doi: 10.1016/S0140-6736(14)61793-1
[69] Sarpel D & Dieterich DT. C-WORTHY: the beginning of the rise of elbasvir and grazoprevir for the treatment of hepatitis C genotype 1 mono and HIV co-infected patients. Ann Transl Med 2016;4(S1):S12–S12. doi: 10.21037/atm.2016.09.22
[70] Zeuzem S, Ghalib R, Reddy KR et al. Grazoprevir-elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med 2015;163(1):1–13. doi: 10.7326/M15-0785
[71] Lawitz E, Gane E, Pearlman B et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase II trial. Lancet 2015.;385(9973):1075–1086. doi: 10.1016/S0140-6736(14)61795-5
[72] Kwo P, Gane E, Peng C-Y et al. P0886: Efficacy and safety of grazoprevir/elbasvir +/- RBV for 12 weeks in patients with HCV G1 or G4 infection who previously failed peginterferon/RBV: C-edge treatment-experienced trial. J Hepatol 2015;62:S674–675. doi: 10.1016/S0168-8278(15)31088-6
[73] Ferenci P, Bernstein D, Lalezari J et al. ABT-450/r–ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med 2014;370(21):1983–1992. doi: 10.1056/NEJMoa1402338
[74] Poordad F, Hezode C, Trinh R et al. ABT-450/r–ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med 2014;370(21):1973–1982. doi: 10.1056/NEJMoa1402869
[75] Hézode C, Asselah T, Reddy KR et al. Ombitasvir plus paritaprevir plus ritonavir with or without ribavirin in treatment-naive and treatment-experienced patients with genotype 4 chronic hepatitis C virus infection (PEARL-I): a randomised, open-label trial. Lancet 2015;385(9986):2502–2509. doi: 10.1016/S0140-6736(15)60159-3
[76] Waked I, Shiha G, Qaqish RB et al. Ombitasvir, paritaprevir, and ritonavir plus ribavirin for chronic hepatitis C virus genotype 4 infection in Egyptian patients with or without compensated cirrhosis (AGATE-II): a multicentre, phase III, partly randomised open-label trial. Lancet Gastroenterol Hepatol 2016;1(1):36–44. doi: 10.1016/S2468-1253(16)30002-4
[77] Feld JJ, Jacobson IM, Hézode C et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med 2015;373(27):2599–2607. doi: 10.1056/NEJMoa1512610
[78] Foster GR, Afdhal N, Roberts SK et al. Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N Engl J Med 2015;373(27):2608–2617. doi: 10.1056/NEJMoa1512612
[79] Charlton M, Everson GT, Flamm SL et al. Ledipasvir and sofosbuvir plus ribavirin for treatment of HCV infection in patients with advanced liver disease. Gastroenterology 2015;149(3):649–659. doi: 10.1053/j.gastro.2015.05.010
[80] Manns M, Samuel D, Gane EJ et al. Ledipasvir and sofosbuvir plus ribavirin in patients with genotype 1 or 4 hepatitis C virus infection and advanced liver disease: a multicentre, open-label, randomised, phase II trial. Lancet Infect Dis 2016;16(6):685–697. doi: 10.1016/S1473-3099(16)00052-9
[81] Curry MP, O’Leary JG, Bzowej N et al. Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med 2015;373(27):2618–2628. doi: 10.1056/NEJMoa1512614
[82] Kidney Disease: Improving Global Outcomes (KDIGO). Foreword. Kidney Int 2008;73(109):S1–2. PMID: 18382440
[83] Fabrizi F, Dixit V & Messa P. Impact of hepatitis C on survival in dialysis patients: a link with cardiovascular mortality? J Viral Hepat 2012;19(9):601–607. doi: 10.1111/j.1365-2893.2012.01633.x
[84] Maluf DG, Fisher RA, King AL et al. Hepatitis C virus infection and kidney transplantation: predictors of patient and graft survival. Transplantation 2007;83(7):853–857. doi: 10.1097/01.tp.0000259725.96694.0a
[85] Roth D, Nelson DR, Bruchfeld A et al. Grazoprevir plus elbasvir in treatment-naive and treatment-experienced patients with hepatitis C virus genotype 1 infection and stage 4-5 chronic kidney disease (the C-SURFER study): a combination phase III study. Lancet 2015;386(10003):1537–1545. doi: 10.1016/S0140-6736(15)00349-9
[86] Pockros PJ, Reddy KR, Mantry PS et al. Efficacy of direct-acting antiviral combination for patients with hepatitis C virus genotype 1 infection and severe renal impairment or end-stage renal disease. Gastroenterology 2016;150(7):1590–1598. doi: 10.1053/j.gastro.2016.02.078
[87] Berenguer J, Ãlvarez-Pellicer J, MartÃn PM et al. Sustained virological response to interferon plus ribavirin reduces liver-related complications and mortality in patients coinfected with human immunodeficiency virus and hepatitis C virus. Hepatology 2009;50(2):407–413. doi: 10.1002/hep.23020
[88] Limketkai BN, Mehta SH, Sutcliffe CG et al. Relationship of liver disease stage and antiviral therapy with liver-related events and death in adults coinfected with HIV/HCV. JAMA 2012;308(4):370–378. doi: 10.1001/jama.2012.7844
[89] Mira JA, Rivero-Juárez A, López-Cortés LF et al. Benefits from sustained virologic response to pegylated interferon plus ribavirin in HIV/hepatitis C virus-coinfected patients with compensated cirrhosis. Clin Infect Dis 2013;56(11):1646–1653. doi: 10.1093/cid/cit103
[90] Naggie S, Cooper C, Saag M et al. Ledipasvir and sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med 2015;373(8):705–713. doi: 10.1056/NEJMoa1501315
[91] Rockstroh JK, Nelson M, Katlama C et al. Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial. Lancet HIV 2015;2(8):e319–327. doi: 10.1016/S2352-3018(15)00114-9
[92] Wyles DL, Ruane PJ, Sulkowski MS et al. Daclatasvir plus sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med 2015;373(8):714–725. doi: 10.1056/NEJMoa1503153
[93] Wyles DL, Brau N, Kottilil S et al. Sofosbuvir and velpatasvir for the treatment of HCV in patients coinfected with HCV and HIV-1: An open-label, phase III study. Clin Infect Dis 2017 Mar [epub ahead of print] doi: 10.1093/cid/cix260
[94] Dieterich D, Bacon B, Curry M et al. Ledipasvir/sofosbuvir+/−ribavirin in patients co-infected with HCV and HIV: real-world heterogeneous population from the Trio Network. J Hepatol 2016;64(2):S756–757. doi: 10.1016/S0168-8278(16)01474-4
[95] Ingiliz P, Christensen S, Kimhofer T et al. Sofosbuvir and ledipasvir for 8 weeks for the treatment of chronic hepatitis C virus (HCV) infection in HCV-monoinfected and HIV-HCV–coinfected individuals: Results from the German hepatitis C cohort (GECCO-01). Clin Infect Dis 2016;63(10):1320–1324. doi: 10.1093/cid/ciw567
[96] Lawitz E, Reau N, Hinestrosa F et al. Efficacy of sofosbuvir, velpatasvir, and GS-9857 in patients with genotype 1 hepatitis C virus infection in an open-label, phase II trial. Gastroenterology 2016;151(5):893–901.e1. doi: 10.1053/j.gastro.2016.07.039
[97] Ng T, Krishnan P, Pilot-Matias T et al. In vitro antiviral activity and resistance profile of the next generation hepatitis C virus NS5A inhibitor pibrentasvir. Antimicrob Agents Chemother 2017;61(5). doi: 10.1128/AAC.02558-16
[98] Poordad F, Felizarta F, Wang S et al. High SVR rates with the combination of ABT-493 + ABT-530 for 8 weeks in non-cirrhotic patients with HCV genotype 1 or 2 infection. Gastroenterology 2016;150(4):S1046–1047. doi: 10.1016/S0016-5085(16)33537-5
[99] Gane E, Poordad F, Wang S et al. High efficacy of ABT-493 and ABT-530 treatment in patients with HCV genotype 1 or 3 infection and compensated cirrhosis. Gastroenterology 2016;151(4):651–659.e1. doi: 10.1053/j.gastro.2016.07.020