Abstract
Cystic fibrosis (CF) is a progressive genetic disorder that affects multiple organs in the body. There are more than 2,000 variants in the CF transmembrane conductance regulator (CFTR) gene. CFTR modulators are small molecules that specifically target the consequences of CFTR gene variants and are the first widely available disease-modifying drugs for people with CF. Ivacaftor was the first CFTR modulator to be licensed in 2012 for people with CF with selected gene variants, but fewer than 10% of people with CF were eligible. In recent years, other CFTR modulators — elexacaftor, lumacaftor and tezacaftor — have become available and now around 90% of people with CF can be treated with these medications. This article gives an overview of the CFTR modulator combinations currently available, real-world clinical outcomes and safety data post-licensing, and discusses the impact that these treatments have had on people with CF. The essential role of the specialist CF pharmacist in the optimisation of these medicines is highlighted throughout, including in the management of drug interactions, adherence and patient access.
Key words: Cystic fibrosis (CF); ivacaftor; elexacaftor; lumacaftor; tezacaftor; cystic fibrosis transmembrane regulator (CFTR); modulator; fertility; adherence; pharmacist.
Key points box:
- Cystic fibrosis (CF) is a genetic disorder with more than 2,000 variants that affects multiple organs causing lung disease, CF-related diabetes, liver dysfunction and more;
- Highly effective medicines called CF transmembrane conductance regulator (CFTR) modulators that target the underlying cause of CF are now available for around 90% of people with CF;
- The CF specialist pharmacist has a crucial role in providing access to modulators, advising on interactions and monitoring liver function and adverse effects;
- Long-term real-world studies will help inform future care for eligible patients;
- Research is ongoing for those with CF who are not eligible for or cannot tolerate a CFTR modulator.
Introduction
Cystic fibrosis (CF) is an autosomal recessive genetic disorder, resulting from the defective CF transmembrane conductance regulator (CFTR) gene. More than 10,000 people in the UK have CF and it is estimated that 1 in 25 people are carriers of this defective gene[1,2]. The CFTR gene encodes the CFTR protein that functions as a dynamic channel that opens and closes to transport anions, predominantly chloride and bicarbonate (and therefore water), at the cell surface[3]. These channels are found in the apical membrane of epithelial cells throughout the body. CFTR gene defects can affect each step of protein synthesis, from errors in transcription, protein translation, folding and trafficking, to expression and gating of the channel on the cell surface, ultimately leading to a reduction in the quantity and/or function of CFTR channels at the cell surface (see Figure 1)[4].
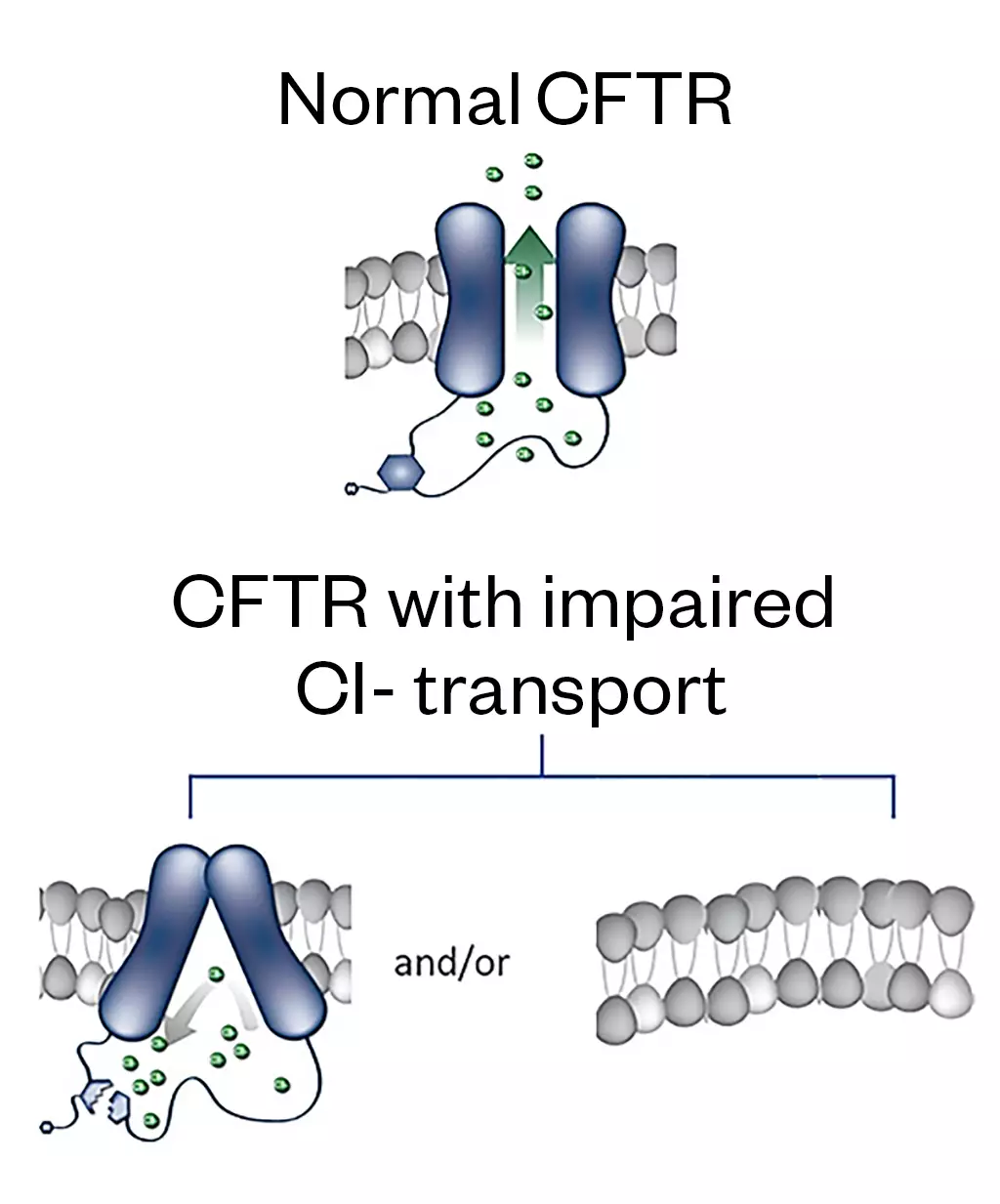
This loss of CFTR protein activity has a significant impact on the ionic balance and hydration of secretions in multiple organs, including the lung, liver, pancreas, digestive tract, reproductive tract and skin. The manifestations of this in various organs include lung disease, which is characterised by chronic infection, leading to inflammation and a progressive decline in lung function; pancreatic insufficiency; CF-related diabetes; liver dysfunction; meconium ileus; raised sweat chloride; and infertility[5].
Pharmacogenomic basis for the use of CFTR modulators
There are more than 2,000 variants in the CFTR gene; as a recessive disorder two copies of a variant will cause CFTR protein dysfunction[6]. More than 350 variants have been classified as disease causing[6]. Many of the variants are rare, occurring in less than 1% of people with CF[6]. The most common variant in the UK CF population is F508del, with 90% of people with CF possessing at least one copy of this variant[1]. People with CF that have two copies of the same genetic variant are described as having a homozygous genotype and those with different variants as heterozygous. Different genetic variants affect CFTR protein activity in differing ways, which is broadly classified into seven classes (see Figure 2[7]).
People with CF who have variants in classes I to III have more clinical manifestations of CF (e.g. pancreatic insufficiency and more frequent infective exacerbations), which leads to a greater decline in the spirometric measure forced expiratory volume in one second (FEV1). A decline in FEV1 is associated with morbidity and mortality in people with CF; therefore, these classes have a poorer prognosis than class III to VI variants[8]. It is important to note that many variants cause CFTR protein dysfunction in more than one way. This classification has defined therapeutic targets and aided the development of disease-modifying treatments for people with CF.
CFTR modulators are small molecules that specifically target the consequences of CFTR gene variants and are the first widely available disease-modifying drugs for people with CF[9]. They represent the most significant breakthrough in the care of people with CF in recent times and are a notable example of the application of personalised medicine.
With the advent of these medicines and increasing prescribing complexities, there is a greater role for the specialist CF pharmacist in the management of people with CF. The specialist CF pharmacist is already an established part of the CF multidisciplinary team and included in the National Service Specification for Cystic Fibrosis[10–12]. Pharmacy provision within this is defined by the Pharmacy Standards of Care produced in conjunction with the Cystic Fibrosis Trust, which are currently being updated[13]. These include a framework for medicines optimisation activities and outline the role of the CF pharmacist and wider pharmacy team.
Overview of current CFTR modulator combinations
An overview of current CFTR modulator combinations can be seen in Table 1; they fall into two categories: potentiators and correctors[4]. Potentiators (i.e ivacaftor), facilitate increased anion transport by potentiating the channel-open probability of the CFTR protein at the cell surface[4]. Correctors facilitate increased anion transport by correcting misfolding errors and increasing the quantity of protein delivered to the cell surface and are used in combination with potentiators to synergistically enhance anion transport of F508del-CFTR protein[4]. The CFTR modulator ‘corrector’ compounds that exert their effect in this way are lumacaftor, tezacaftor and elexacaftor[4]. The action of this group of drugs is important for people with CF who are homozygous or heterozygous for the most common variant in people with CF in the UK — F508del; a class II variant that results in a misfolding of the CFTR protein and reduced delivery to the cell surface[1].
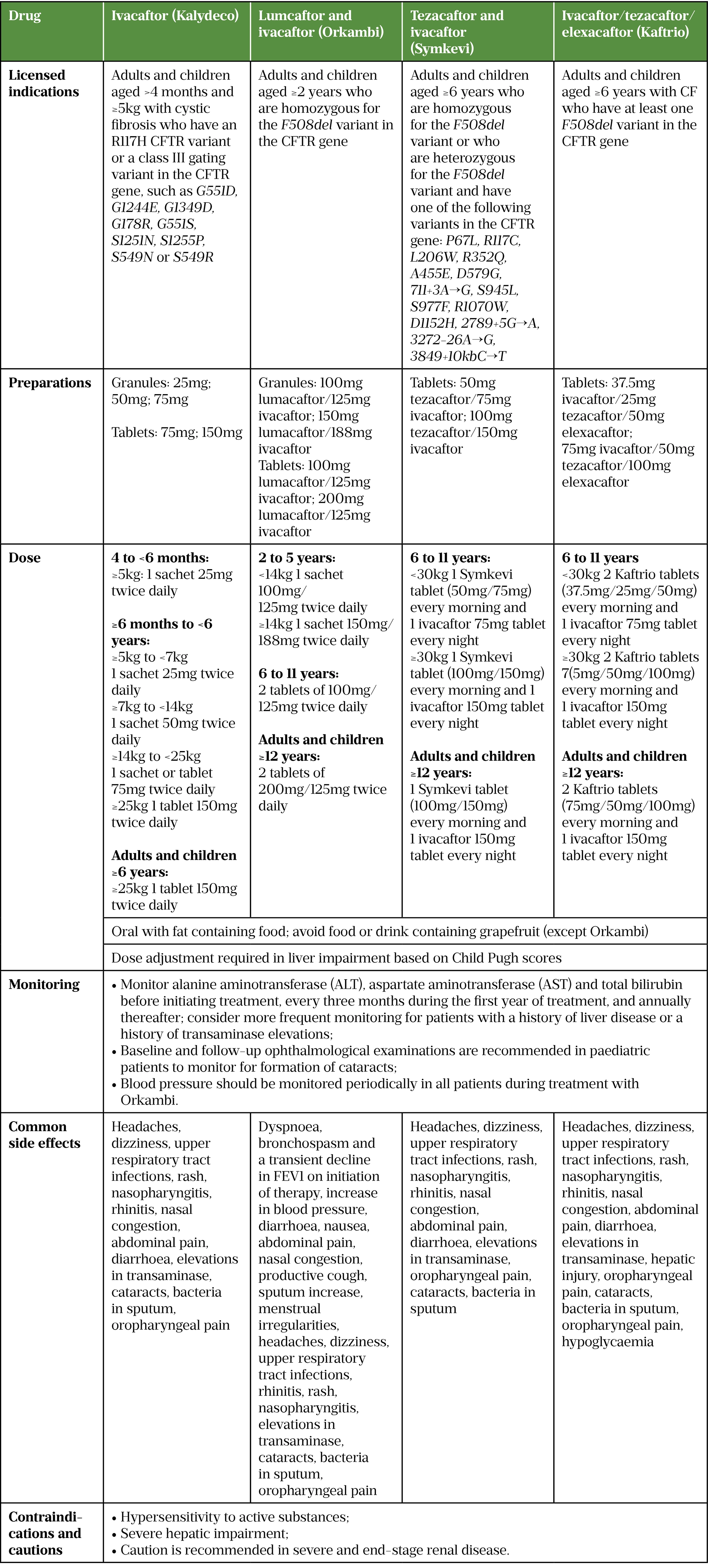
At the time of publication, ivacaftor is available as a single agent (Kalydeco; Vertex Pharmaceuticals)[14]. Combination products available in the UK consisting of potentiator and corrector compounds are: ivacaftor + lumacaftor (Orkambi; Vertex Pharmaceuticals); ivacaftor + tezacaftor (Symkevi; Vertex Pharmaceuticals); and the triple combination elexacaftor + ivacaftor + tezacaftor (Kaftrio; Vertex Pharmaceuticals)[15–17].
Ivacaftor
In 2012, ivacaftor was the first CFTR modulator to be licensed for use in people with CF, as monotherapy for people with class III (gating) variants and was commissioned for use in the UK in 2013[14,18]. Gating variants, the most common being G551D, result in adequate delivery of protein to the cell surface but incorrect opening of the channel. Ivacaftor may also benefit people with class IV variants in which the channels at the surface open properly but anion conductance is reduced and for class V variants, which result in reduced protein transcription and a reduced quantity of the protein being produced[19].
The first phase III study of ivacaftor in adults and adolescents aged 12 years and over — STRIVE — demonstrated an impressive improvement in lung function at week 48 (increase in FEV1 % predicted of 10.5% compared to placebo; P<0.001) as well as a 55% reduction in the risk of a pulmonary exacerbation (P<0.001); and improvements in weight, sweat chloride (as a measure of CFTR activity), and patient-reported respiratory symptoms as assessed using the respiratory domain of the ‘CF questionnaire – revised’ (CFQ-R)[20,21]. The incidence of adverse events was similar between the two groups. Similar results were seen in the ENVISION study, comparing ivacaftor with a placebo in children between the ages of 6 and 11 years[22]. An open label, roll-over continuation study — PERSIST — also showed that the improvement in predicted FEV1 %, weight gain and pulmonary exacerbation rate were sustained at 144 weeks[23].
R117H is a class IV variant; however, it is also known to display a defect in channel gating[24]. Consequent studies in individuals with this variant showed that although there was no significant improvement in FEV1% predicted, ivacaftor did significantly improve sweat chloride and CFQ-R respiratory domain score and was subsequently licensed for this variant[14,25].
The focus of studies in infants and younger children aged under six years old, such as KIWI, ARRIVAL and KLIMB, have demonstrated the safety, pharmacokinetics, and pharmacodynamics of ivacaftor[26–29]. Improvements were seen in sweat chloride, weight, BMI scores and the biomarker for pancreatic insufficiency faecal elastase-1, with improvements in the latter biomarker suggesting that ivacaftor might preserve pancreatic function if started early in life[28]. Ivacaftor has subsequently been licensed as monotherapy for infants and children from four months of age with gating variants[30] (see Table 1).
Lumacaftor + ivacaftor
In 2019, lumacaftor and ivacaftor became the first combination therapy approved for use in the UK, specifically targeting those who are homozygous for the F508del variant[31]. This is the most common variant in the UK, with a prevalence of 47.7%, and hugely increased the number of people with CF eligible for modulators[1]. People with this variant have a reduced amount of the CFTR protein at the cell surface so ivacaftor alone is not sufficient to significantly improve CFTR function[6].
The licence for lumacaftor + ivacaftor was based on two phase III studies — TRAFFIC and TRANSPORT — which compared it to a placebo in people with CF aged 12 years and over, who were homozygous for F508del[32]. In contrast to ivacaftor, only modest improvements of 2.8% (P<0.001) were seen in FEV1% predicted, alongside an increased BMI (+0.26kg/m2) and statistically significant but modest improvements in a validated symptom score (CFQ-R). Similar to ivacaftor, the combination was well tolerated, with the most significant side effect being elevation of liver function tests. However, raised blood pressure, dyspnoea and chest tightness — mainly on initiation of therapy — were reported more frequently in the active treatment groups[32].
The effects of lumacaftor + ivacaftor were comparable in children aged 6 to 11 years, with similar changes in FEV1% predicted observed, and a comparable safety profile[33]. The safety and pharmacokinetics of lumacaftor + ivacaftor were investigated in an open label study in children aged 2 to 5 years and were found to be consistent with studies in older children and adults[34]. Mean sweat chloride concentrations were significantly reduced and, alongside improvements in growth parameters and biomarkers of pancreatic function at 24 weeks, the evidence for early introduction of CFTR modulators to attenuate the deleterious effects of CFTR dysfunction was expanded. Lumacaftor + ivacaftor is now licensed in children and adults aged from 2 years[15]. An open-label study in children between 12 and 24 months old will further investigate the safety and tolerability (NCT04235140)[35].
Tezacaftor + ivacaftor
In 2018, a further dual combination of tezacaftor with ivacaftor was approved for use in the UK, which sought to build on the limited efficacy of lumacaftor + ivacaftor and further broaden the population for which CFTR modulators were available. In EVOLVE, a randomised, double-blind, multicentre, placebo-controlled trial of tezacaftor + ivacaftor in people with CF aged 12 years and older who were homozygous for the F508del variant, it was associated with a significantly lower frequency of pulmonary exacerbations and improvement in FEV1% predicted of 3.4% compared to baseline; a magnitude of effect similar to that seen with ivacaftor + lumacaftor[36].
Tezacaftor + ivacaftor was investigated in CF patients heterozygous for F508del and either a residual function variant (class IV or V) in EXPAND or minimal function variant (class I, II or III)[37]. EXPAND demonstrated an average absolute improvement in FEV1% predicted of 6.8% compared with the placebo; however, no significant clinical benefit was seen in patients with F508del and a minimal function genotype[38]. In children aged 6 to 11 years who were homozygous for F508del or heterozygous for F508del with a residual function variant, the pharmacokinetics, safety, tolerability and effect on CFTR function of ivacaftor + tezacaftor was consistent with that seen in older children and adults[39]. Furthermore, a study utilising lung clearance index (LCI) as a more sensitive measure of lung function in younger children showed a significant improvement in LCI for those on tezacaftor + ivacaftor[40]. Ivacaftor + tezacaftor is licensed for people with eligible genotypes aged six years and above[16].
Elexacaftor + tezacaftor + ivacaftor
The limited efficacy of both dual combination therapies led to the development of the triple combination with the addition of another corrector — elexacaftor — to the dual tezacaftor + ivacaftor combination. This demonstrated an impressive improvement in FEV1% predicted of 10% at four weeks versus ivacaftor / tezacaftor, where a 3.4% increase in FEV1% predicted had already been demonstrated versus placebo, in people aged 12 years and older homozygous for F508del[24,41]. This was comparable to the increase seen in people heterozygous for F508del with a minimal function variant, where an improvement in FEV1% predicted of 14.3% was seen at 24 weeks versus placebo[42]. Moreover, the triple combination led to a 63% reduction in pulmonary exacerbations per year and a 20-point improvement in CFQ-R respiratory domain score versus placebo in F508del heterozygotes[42]. This proved to be significant for this cohort of patients, in whom previous CFTR modulators were less effective.
The most recent study included individuals 12 years and older with one copy of F508del variant and a residual function or gating variant, versus ivacaftor or ivacaftor + tezacaftor[43]. An additional improvement in FEV1% predicted of 3.5% was seen in the elexacaftor + tezacaftor + ivacaftor group relative to the active control groups. The triple combination also showed good tolerability with low rates of discontinuation.
An open-label study in children aged 6 to 11 years who were either homozygous for F508del or heterozygous for F508del with a minimal function variant, established the safety and tolerability of elexacaftor + tezacaftor + ivacaftor for this age group, along with an increase in FEV1% predicted of 10.2% from baseline, improvements in LCI, sweat chloride, CFQ-R respiratory domain score and BMI-for-age z-score at 24 weeks[44]. A similar study in children aged 2 to 5 years old is ongoing (NCT04537793) and given the impressive effects seen thus far in older children and adults, the results are eagerly awaited[45].
In 2020, elexacaftor + tezacaftor + ivacaftor was licensed and approved for use in the UK for adults and children over 12 years of age who have at least one copy of the F508del variant[46]. It was made available to children aged 6 years and over in the UK in January 2022, and thus expanded the accessibility of highly effective modulator therapy for a large proportion of people with CF[17,47].
Patient access to CFTR modulators
In the UK, CFTR modulators are commissioned for use in people with CF and access agreements between the manufacturer and the relevant health bodies within the UK have allowed extensions to licensed indications to be commissioned and made available to patients as soon as they have been approved by the Medicines and Healthcare products Regulatory Agency (MHRA)[48–51]. The commissioning agreements in place also include the use of CFTR modulators for ‘off-label’ indications. These indications include people with CF who have rare variants for which there is in vitro data that indicate the variant to be responsive to available modulator compounds, but which might not have been otherwise included in clinical studies owing to their rarity[48]. This is in line with the approach taken in the United States by the US Food and Drug Administration[52].
There remains a subset of people with CF — about 10% of the CF population — for whom there is no approved modulator available. Moreover, research has shown that people with CF from ethnic minority backgrounds were least likely to be eligible for a modulator owing to the rarity of their variants[53,54]. Class I and VII variants have limited therapeutic options[6]. Class VII variants lead to an absence of messenger RNA and, to date, a therapeutic target has not been identified. Class I variants are called ‘nonsense’ or ‘stop’ variants because they are usually premature terminating codons, which result in shortened dysfunctional CFTR protein being produced. Drugs known as ‘read-through agents’, which allow ribosomes to read through stop codons to produce functioning CFTR protein, have the potential to be used for people with these variants.
For example, ataluren, a drug licensed for Duchenne muscular dystrophy — a condition that arises from a nonsense variant — was investigated for use in CF patients with class I variants. Unfortunately, study results have showed no statistical difference in lung function or pulmonary exacerbation rate in those patients given ataluren versus placebo over 48 weeks[55]. However, there are several other read-through agents that are in the early phases of the clinical trials pipeline that may address this gap[56]. For people with rare variants who might otherwise not be included in drug trials for currently available modulators, the ‘HIT-CF Europe’ study uses an in vitro ‘organoid’ system. Organoids are cells cultured from rectal tissue samples from people with CF[57]. Because organoids are made from stem cells, they contain the same variants as the person from whom the tissue samples are derived. CFTR modulators in development can then be tested on these organoids to assess drug response for a particular patient[58]. The ultimate goal of this study is to “develop a path for access to therapies for individual patients or patient groups who show positive response to the therapy in an organoid test and pave the way for organoid-based personalized medicine”[59].
Given that the detrimental effects of CFTR protein dysfunction begin from birth, and likely in utero, it is desirable that disease-modifying agents are commenced as early as possible, with antenatal treatment not an unreasonable aim for the future[60,61]. However, of the available modulators, only ivacaftor has evidence supporting its use in infants down to four months of age for a relatively small section of the paediatric CF population who have gating variants[29]. It is hoped that the ongoing open-label elexacaftor + tezacaftor + ivacaftor study, which is being carried out in children aged between two and five years who are homozygous or heterozygous for F508del (NCT04537793), will bridge the gap between the availability of highly effective modulators for a larger proportion of infants and children with CF[45].
It should also be recognised that the high cost of CFTR modulators has widened the disparity in CF care across the world, with many developing nations unable to afford them. Though the price recognises the significant amount of investment that is required to bring new medicines to market, there is a pressing need to work across boundaries, including with the pharmaceutical industry and governments to provide affordable access to CFTR modulators for all people with CF globally[10].
‘Real-world’ clinical outcomes
There is a great deal of published real world clinical experience about ivacaftor, the modulator that has been clinically available for the longest. Many studies have shown a reduction in pulmonary exacerbations requiring treatment with antibiotics, increase in BMI, improvement in control of CF-related diabetes and reduction in need for pancreatic enzyme replacement therapy[62]. Some studies have shown after an acute improvement in lung function, FEV1 declines and, at five years, has returned to pre-ivacaftor levels[63–65].
There are still limited real-world data available on the effect of the newer CFTR modulators on clinical outcomes[66]. Several prospective, multicentre, longitudinal observational studies that will measure the impact of modulators on a wide range of CF disease manifestations and organ systems are underway in Europe and the United States. In addition to providing safety and efficacy data beyond phase II and III trials, studies that include children <12 years will deliver information about the long-term effect of using CFTR modulators in infants and young children (PROMISE NCT04613128; PROMISE Paeds NCT03808376; RECOVER NCT04602468; BEGIN NCT04509050)[67–70].
‘Real-world’ safety
As with the data on the real-world effects of modulators on clinical outcomes, knowledge regarding the real-world safety of modulators is largely limited to ivacaftor and ivacaftor + lumacaftor. A recent systematic review found that the side effect profile of these modulators is in line with that seen in clinical studies. Dyspnoea and chest tightness resulting in discontinuation of ivacaftor + lumacaftor was more prevalent in real-world studies than in phase III studies; this is thought to be caused by lumacaftor, as similar effects were not observed with ivacaftor monotherapy. Rash was also a commonly reported side effect, although this rarely required intervention[71].
Hepatic side effects are synonymous with all CFTR modulators and were reported in all phase III studies of these medicines. Monitoring of liver function tests and dose adjustments are recommended for liver impairment based on Child Pugh scores[14–17]. Following a European review of safety data on elexacaftor + ivacaftor + tezacaftor and reports to the MHRA via the Yellow Card Scheme, the MHRA issued an alert in February 2022 regarding the risk of serious liver injury from elexacaftor + ivacaftor + tezacaftor[72]. This strengthened previous recommendations on monitoring liver function to advise measuring total bilirubin concomitantly. The advice that more frequent monitoring should be considered in patients with a history of transaminase elevations was also extended to include patients with pre-existing liver disease[72].
People with CF experience high levels of anxiety and depression, as may be expected with a chronic, complex disease[73]. Despite depression and anxiety not being reported as an adverse event in the clinical trials involving CFTR modulators, there have been reports in the literature. Patients taking ivacaftor + lumacaftor have experienced new onset of depression, worsening of existing depression and bipolar disorder associated with suicidal ideation[74–77]. In a case series of 44 people with CF who switched from ivacaftor+ lumacaftor to ivacaftor + tezacaftor, 5 out of the 44 reported neurocognitive side effects including sleep disturbance, depersonalisation and visual hallucination[66].
In 2022, Heo et al. published a case series of six patients who reported mental fogginess, memory issues and insomnia; three of these patients were able to stay on modulator treatment by adjusting dosing schedules and one person found that their symptoms improved over time[77]. Though there are similar anecdotal reports of neurocognitive side effects in elexacaftor + ivacaftor + tezacaftor, this is confounded by its commissioning during the COVID-19 pandemic and the wider psychological impact of this unparalled time. More data on psychological adverse effects of CFTR modulators are likely to emerge with time. People with CF should have access to a CF specialist psychologist via their CF centre and it is important that if people are experiencing neuropsychological issues with their modulator, they discuss this with their CF team.
Effects on weight
Studies have shown that ivacaftor has a positive impact on nutritional status as well as lung function, with an increase in BMI in both adults and paediatric cohorts[64,78]. Trial data suggest that elexacaftor + tezacaftor + ivacaftor increases BMI and this is also being seen in real-world studies[41,42,79]. In 2022, results from an online survey carried out by UK-wide CF dietitians show that 77% of respondents’ weight had increased and that 31% had gained at least 10kg[80]. This finding has been borne out by other groups and nutritional management of people with CF who are taking CFTR modulators has shifted from advising a high-calorie and high-fat diet to advice on how to manage weight gain and prevent obesity[81–83]. Anecdotal evidence has shown that a small number of people are choosing to switch modulator from elexacaftor + ivacaftor + tezacaftor to ivacaftor monotherapy or ivacaftor + tezacaftor owing to unacceptable weight gain.
Effects on fertility
Prevalence of infertility or subfertility in women with CF has been estimated to be 20–35%; the reasons for this are not fully understood[84]. Epithelial cells in the cervix are impacted by CFTR gene variants, resulting in thick cervical mucus and a pH-imbalance, which may impact pregnancy outcomes.
Pregnancy rates in women with CF have been increasing since the introduction of ivacaftor[85]. With the advent of elexacaftor + ivacaftor + tezacaftor and the possibility of increased life expectancy, more women with CF will be able to have a family[85]. There are an increasing amount of data on the safe use of CFTR modulators and data to support the continuation of modulator therapy to prevent clinical decline during pregnancy[85–88]. The MAYFLOWERS trial is a multi-centre observational study that will follow pregnant women with CF, conducted in 40 CF centres in the United States. Women will be enrolled during their first trimester and assessed at regular intervals until two years after delivery. Changes in lung function over the course of pregnancy will be evaluated based on CFTR modulator use (NCT04828382)[89].
When counselling females of child-bearing age with CF on elexacaftor + ivacaftor + tezacaftor, the CF specialist pharmacist should ensure that they are aware of the potential for increased fertility and the need for adequate contraception if appropriate.
Impact on adherence
Adherence to CF medicines is reportedly low, especially with more burdensome medicines, such as nebulised antibiotics and mucolytics; however, adherence to CFTR modulators is expected to be higher owing to ease of administration and demonstrable patient benefits[90]. In 2018, Kirkham et al. reported ivacaftor adherence rates of 56–68% (SD 28%) over a 12-month period[91]. In 2015, Siracusa et al. showed that ivacaftor had a mean adherence of 61% (SD, 28%) in 12 patients followed up for a mean of 118 days, with adherence declining over time, by 1.9% per 8 weeks[92]. Then, in 2020, Olivereau et al. found mean adherence rates for 96 people on ivacaftor + lumacaftor of 89% at 6 months and 83% at 12 months[93].
In 2021, Mehta et al. used a national specialty pharmacy database to calculate adherence to ivacaftor, ivacaftor + lumacaftor and ivacaftor + tezacaftor in 2,548 patients, with results showing that the proportion of days covered (a measurement of adherence similar to medicines possession ratio; MPR) of 0.92 for ivacaftor + tezacaftor and 0.84 for the other modulators[94]. Meanwhile, Marrero Alvarez et al. found that 98.7% (n=81) patients on ivacaftor + tezacaftor, elexacaftor + ivacaftor + tezacaftor or ivacaftor + lumacaftor had adequate adherence, defined as a MPR of ≥80%[95].
Conversely, the therapeutic benefits of CFTR modulators have been found to adversely affect adherence to other CF treatments, as the adverse consequences of CF are reduced in most people taking modulators. Howell et al. found that 65% of patient’s adherence to nebulised therapies reduced after starting elexacaftor + ivacaftor + tezacaftor, while mean nebulised adherence therapy dropped from 65% to 42%[96]. It should be noted that clinical trials of modulator therapy were carried out in patients as an additional therapy to routine CF medications. It is therefore important to investigate whether the same clinical benefits can be derived without key parts of the regimen, such as mucolytics, particularly in lieu of this emerging adherence data.
This highlights a top research priority identified for people with CF, which looks at reducing or simplifying the treatment burden[97]. A registry-based trial investigating therapy rationalisation in people with CF (CF STORM) is a prospective, randomised, open-label, two-arm non-inferiority trial that is aimed to investigate if patients aged 12 years and over who are established on elexacaftor + ivacaftor + tezacaftor can safely stop taking nebulised mucoactive drugs without a significant fall in their respiratory function[98]. Secondary outcome measures are looking at change in antibiotic use, quality of life and weight[98].
SIMPLIFY is a very similar study being carried out in the United States investigating discontinuation of the same medications with the same primary objective and similar secondary outcome measures including incidence of adverse events (NCT04378153)[99]. The ‘Home-Reported Outcomes in People with Cystic Fibrosis taking highly effective CFTR modulator therapy’ (HERO) trial is a prospective, observational cohort study being carried out in the United States in people with CF aged over 12 years, which is investigating changes in lung function in people who make at least one change to their chronic daily therapies compared to people who have not made any changes (NCT04798014)[100].
Though the outcomes of these studies will be vital when defining how modulator therapy may impact the overall treatment burden for a person with CF, the CF specialist pharmacist can work with people with CF and their families to support and encourage them to take their medicines as prescribed. Research that is investigating modifications to existing compounds, such as deuterated ivacaftor, to provide a once-daily dosing regimen may also be crucial in supporting adherence (NCT03911713)[101]. This is vital given the high cost to the NHS, including not just the cost of the drugs themselves, but the consequences of not taking treatment as intended (e.g., hospital admissions for IV antibiotics).
Managing CFTR modulator interactions
Elexacaftor, ivacaftor and tezacaftor are primarily metabolised by the cytochrome P450 pathway, specifically the enzymes CYP3A4 and CYP3A5[14,16,17]. Because these drugs — particularly ivacaftor — are substrates for these enzymes, there are many interactions with other medicines commonly used in people with CF (see Table 2). Lumacaftor is not metabolised via the CYP450 system; however, it is a strong inducer of CYP3A4, CYP2CP, CYP2C19 and CYP2B6, and when combined with ivacaftor, which is a weak inhibitor of CYP3A4 and CYP2C19, the overall effect is still strong induction of these enzymes[15]. This means that additional consideration is needed when using lumacaftor + ivacaftor in combination with more medicines, more dose adjustments are required and risk of treatment failure with certain drug combinations is higher than with the other modulators.
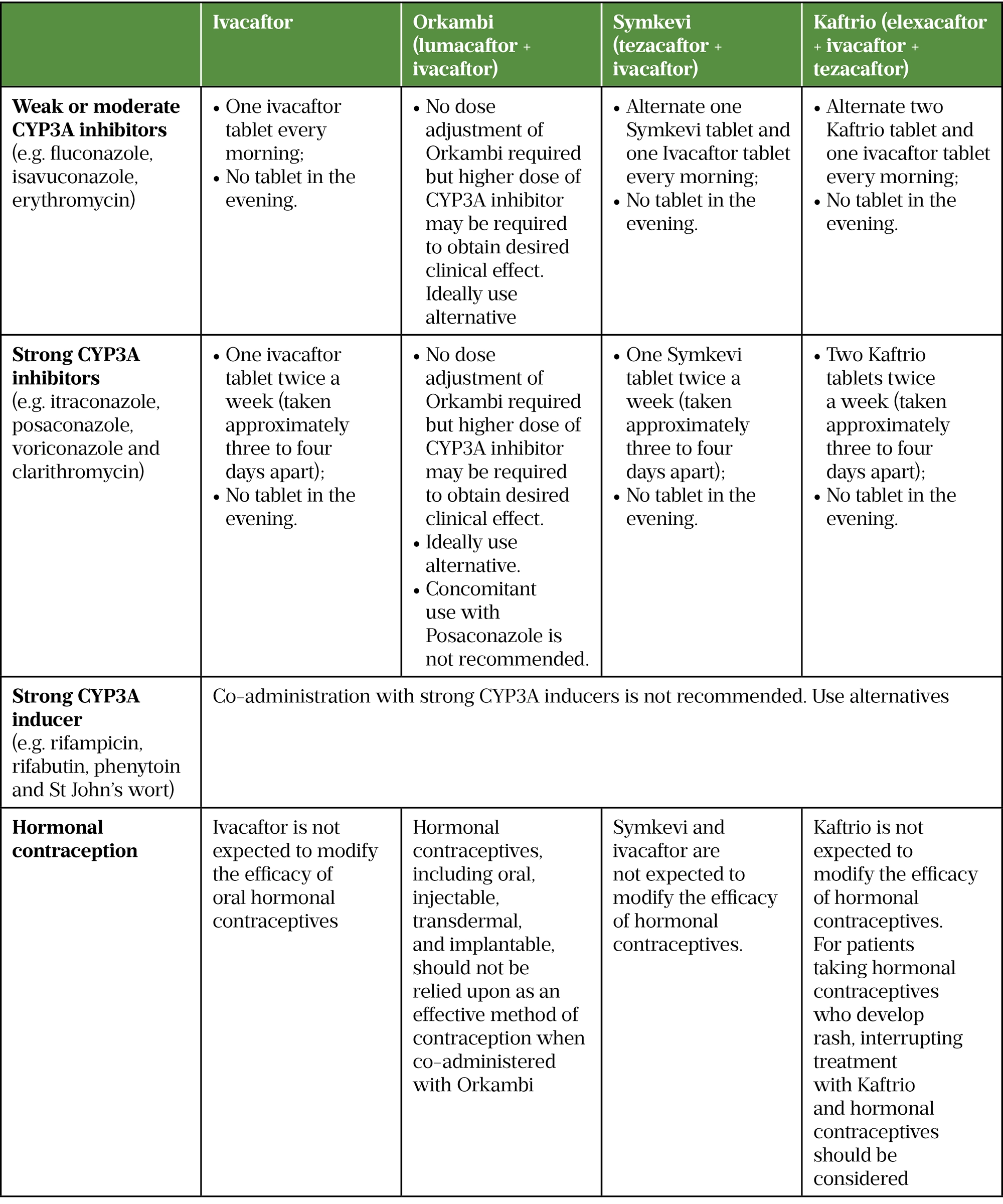
During 2020, 17.2% of people with CF in the UK grew Aspergillus in their sputum and 8.2% grew non-tuberculous mycobacteria (NTM)[1]. These organisms often require prolonged courses of antifungals or combinations of antibiotics and managing interactions between CFTR modulators and medicines required to treat these organisms can be challenging[102]. For example, people taking azole antifungals need to reduce the dose of their modulator depending on the azole (see Table 2), and the choice of antibiotics used to treat NTM is limited by not being able to use rifampicin, while modulator dose reduction is required when taking long-term clarithromycin[14–17]. Once these treatments are stopped, a washout period is needed before full-dose modulator treatment can be restarted. To further complicate matters, it is recognised that there is inter- and intra-individual variability in the metabolism of medicines and there is a risk of underdosing CFTR modulators when using ‘blanket recommendations’ with the potential for reduced effectiveness[103,104]. The ability to measure plasma levels of individual modulators may help to guide dosage adjustments but assays to enable this are not currently commercially available. In the absence of this, some centres have chosen to use sweat chloride as a surrogate measure to manage interactions and subsequent dose adjustments[105].
It is important to remember that even with the many positive effects of CFTR modulators on CF disease manifestations, many people with CF will have established irreversible structural lung disease before commencing CFTR modulator therapy and will therefore continue to require adjunctive therapies to manage this[106]. Given the many clinically relevant drug interactions between CFTR modulators and commonly used medicines (including those available over the counter) in CF (see Table 2), this will present an ongoing clinical challenge and is another vital part of the CF specialist pharmacist’s role.
Patient perception of CFTR modulators
The physical impact of CFTR modulators is well documented and easily measurable; however, it is also well recognised that CFTR modulators can have a significant impact on the psychological and psychosocial wellbeing of people with CF. Several studies that have focused on, or have included, psychosocial aspects of life have shown that taking a CFTR modulator improves quality of life[107]. People with CF no longer see their CF diagnosis as their defining feature and are more able to focus on other aspects of their lives, such as relationships and careers[108]. A feeling of hope and optimism about the future was a common feature in one small US study[109]. The Cystic Fibrosis Trust has published blogs by people who no longer cough and have been able to achieve things such as climbing a hill, which they could not have done pre-modulator[110].
Conversely, a common feeling in those with CF eligible for a CFTR modulator is ‘survivor guilt’ — they have an opportunity that others with CF do not have[111]. Those taking modulators are finding their perception of CF and themselves has changed and can struggle with their new identity[111]. If a person with CF has not had the response to a CFTR modulator they were hoping for, or has not been able to tolerate it, they can feel disappointed or even experience a sense of grief[111].
CFTR modulators have had such an impact on the CF community, that the Cystic Fibrosis Trust, in conjunction with healthcare professionals working in CF and people with CF, have produced a factsheet about complex and individual experiences with elexacaftor + ivacaftor + tezacaftor[111].
Future research
Although we celebrate the positive impact that CFTR modulators have had on people with CF, we must not forget people who are not eligible for them. They can feel understandably frustrated, sad, angry and that they have been ‘left behind’[111,112]. Research to develop new CFTR disease-modifying therapies for those not currently eligible for modulators is crucial. Equally, ongoing research into use of adjunctive therapies to manage the manifestations of CF is still required for this group, and for those who may not be able to tolerate modulators in the long term. Gene therapy — the introduction of a functional gene into host cells to replace a defective gene — has been investigated as a potential therapy for CF on several occasions since the CFTR gene was discovered in 1989 and may provide an option for patients who are ineligible (or intolerant) for modulators[113]. Though progress in this area has been hindered by several factors, including difficulties in delivering gene therapy to affected cells and safety concerns around malignancies as a result of off-target insertional mutagenesis, a renewed programme is being established[114,115]. Other genetic therapies with potential include the use of mRNA technology to insert functional mRNA into cells — currently being investigated in the phase 1/2 RESTORE-CF study [NCT03375047] — and gene editing, utilising CRISPR gene editing technology to repair the defective CFTR gene[116,117].
We also must not forget that although CFTR modulators are a significant step towards rectifying the root cause of CF, they are unlikely to completely reverse damage that may have already occurred. Therefore, research into use of CFTR modulators in the youngest age groups — which may prevent this damage in the first place — is paramount and fundamental to maximising the benefits of CFTR modulators for people with CF.
Conclusion
The development and introduction of CFTR modulators over the past decade has transformed the lives of many people with CF. The recently expanded eligibility criteria, improving the availability of modulator therapy for an even larger cohort of people with CF, alongside the encouraging ‘real-world’ outcomes that we are seeing in patients taking ivacaftor means we can expect the therapeutic landscape for many people with CF to continue to evolve over the coming decades. Though the impact of CFTR modulators on life expectancy in people with CF is still to be fully evaluated, it can safely be assumed that life expectancy will increase[118]. The Cystic Fibrosis Trust 2020 registry report, published in December 2021, celebrated predicted survival greater than 50 years for the first time[1].
It is important to consider the ageing population of people with CF that will bring medical complications and challenges that CF teams have not had to deal with previously, such as cardiovascular disease, type 2 diabetes mellitus, liver disease and cancer[119,120]. The risk of colon cancer is five to ten times greater in people with CF compared to the general population[121]. As mentioned earlier, people taking elexacaftor + ivacaftor + tezacaftor are likely to gain weight, which introduces obesity and its effect on morbidity as another challenge for the CF team. CF specialist pharmacists are well placed to manage the increasing pharmaceutical complexity presented by these medicines and the potential for polypharmacy when treating an ageing population.
Additionally, as more women with CF become pregnant, a CF specialist pharmacist can ensure that the medication a woman is taking at conception is safe in pregnancy and advise on any medication that needs to be prescribed during the pregnancy. Working within the CF multidisciplinary team, the CF specialist pharmacist can provide leadership to evaluate and respond to the changing requirements of this cohort of patients to facilitate medicines optimisation and ensure best value use of medicines in line with national commissioning priorities.
Financial disclosures/conflict of interest
There are no financial disclosures or conflicts of interest to declare.
Author contributions
Siân Bentley, Elaine Bowman, Sukeshi Makhecha contributed equally to the development of this manuscript.
Nominate an early-careers researcher today for the OPERA23 award
Help us in our search for outstanding early-career researchers who are accomplishing great things in pharmacy and pharmaceutical science.
For entry criteria and details on how to nominate a colleague, click here.
- 1UK Cystic Fibrosis Registry 2020 Annual Data Report. . Cystic Fibrosis Trust. 2021.https://www.cysticfibrosis.org.uk/sites/default/files/2022-05/2020%20Annual%20data%20report%20-%20Version%204.pdf (accessed Oct 2022).
- 2Cystic fibrosis FAQs. Cystic Fibrosis Trust. https://www.cysticfibrosis.org.uk/what-is-cystic-fibrosis/faqs#How%20common%20is%20cystic%20fibrosis? (accessed Oct 2022).
- 3Stoltz DA, Meyerholz DK, Welsh MJ. Origins of Cystic Fibrosis Lung Disease. N Engl J Med. 2015;372:351–62. doi:10.1056/nejmra1300109
- 4Lopes-Pacheco M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020;10. doi:10.3389/fphar.2019.01662
- 5Hanssens LS, Duchateau J, Casimir GJ. CFTR Protein: Not Just a Chloride Channel? Cells. 2021;10:2844. doi:10.3390/cells10112844
- 6The Clinical and Functional TRanslation of CFTR (CFTR2) . The Clinical and Functional TRanslation of CFTR (CFTR2). https://cftr2.org (accessed Oct 2022).
- 7De Boeck K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020;109:893–9. doi:10.1111/apa.15155
- 8Szczesniak R, Heltshe SL, Stanojevic S, et al. Use of FEV1 in cystic fibrosis epidemiologic studies and clinical trials: A statistical perspective for the clinical researcher. Journal of Cystic Fibrosis. 2017;16:318–26. doi:10.1016/j.jcf.2017.01.002
- 9Roda J, Pinto-Silva C, Silva IAI, et al. New drugs in cystic fibrosis: what has changed in the last decade? Therapeutic Advances in Chronic Disease. 2022;13:204062232210981. doi:10.1177/20406223221098136
- 10Bell SC, Mall MA, Gutierrez H, et al. The future of cystic fibrosis care: a global perspective. The Lancet Respiratory Medicine. 2020;8:65–124. doi:10.1016/s2213-2600(19)30337-6
- 11Service Specification: Cystic Fibrosis – Adults. NHS England. 2018.https://www.england.nhs.uk/wp-content/uploads/2018/08/Cystic-fibrosis-adult.pdf (accessed Oct 2022).
- 12Service Specification: Cystic Fibrosis – Children. NHS England. 2018.https://www.england.nhs.uk/wp-content/uploads/2018/07/a01Sb-spec-cystic-fibrosis-child.pdf (accessed Oct 2022).
- 13Pharmacy Standards in Cystic Fibrosis Care 2011. Cystic Fibrosis Trust. 2011.https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/Pharmacy%20standards%20of%20care.pdf (accessed Oct 2022).
- 14Kalydeco 150 mg Film-coated Tablets. Electronic Medicines Compendium. https://www.medicines.org.uk/emc/product/3040/smpc (accessed Oct 2022).
- 15Orkambi 100 mg/125 mg granules in sachet. Electronic Medicines Compendium. 2022.https://www.medicines.org.uk/emc/product/3040/smpc (accessed Oct 2022).
- 16Symkevi 50 mg/75 mg film coated tablets. Electronic Medicines Compendium. 2022.https://www.medicines.org.uk/emc/product/11971/smpc (accessed Oct 2022).
- 17Kaftrio 37.5mg 25 mg 50 mg film-coated tablets. Electronic Medicines Compendium. 2022.https://www.medicines.org.uk/emc/product/13216/smpc (accessed Oct 2022).
- 18Clinical Commissioning Policy: Ivacaftor for Cystic Fibrosis. NHS England. 2013.https://www.england.nhs.uk/wp-content/uploads/2013/04/a01-p-b.pdf (accessed Oct 2022).
- 19Guigui S, Wang J, Cohen RI. The use of ivacaftor in CFTR mutations resulting in residual functioning protein. Respiratory Medicine Case Reports. 2016;19:193–5. doi:10.1016/j.rmcr.2016.10.012
- 20Ramsey BW, Davies J, McElvaney NG, et al. A CFTR Potentiator in Patients with Cystic Fibrosis and theG551DMutation. N Engl J Med. 2011;365:1663–72. doi:10.1056/nejmoa1105185
- 21Quittner A, Suthoff E, Rendas-Baum R, et al. Effect of ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation: patient-reported outcomes in the STRIVE randomized, controlled trial. Health Qual Life Outcomes. 2015;13. doi:10.1186/s12955-015-0293-6
- 22Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and Safety of Ivacaftor in Patients Aged 6 to 11 Years with Cystic Fibrosis with a G551D Mutation. Am J Respir Crit Care Med. 2013;187:1219–25. doi:10.1164/rccm.201301-0153oc
- 23McKone EF, Borowitz D, Drevinek P, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp- CFTR mutation: a phase 3, open-label extension study (PERSIST). The Lancet Respiratory Medicine. 2014;2:902–10. doi:10.1016/s2213-2600(14)70218-8
- 24Van Goor F, Yu H, Burton B, et al. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. Journal of Cystic Fibrosis. 2014;13:29–36. doi:10.1016/j.jcf.2013.06.008
- 25Moss RB, Flume PA, Elborn JS, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. The Lancet Respiratory Medicine. 2015;3:524–33. doi:10.1016/s2213-2600(15)00201-5
- 26Davies JC, Cunningham S, Harris WT, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. The Lancet Respiratory Medicine. 2016;4:107–15. doi:10.1016/s2213-2600(15)00545-7
- 27Rosenfeld M, Cunningham S, Harris WT, et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2–5 years (KLIMB). Journal of Cystic Fibrosis. 2019;18:838–43. doi:10.1016/j.jcf.2019.03.009
- 28Rosenfeld M, Wainwright CE, Higgins M, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. The Lancet Respiratory Medicine. 2018;6:545–53. doi:10.1016/s2213-2600(18)30202-9
- 29Davies JC, Wainwright CE, Sawicki GS, et al. Ivacaftor in Infants Aged 4 to <12 Months with Cystic Fibrosis and a Gating Mutation. Results of a Two-Part Phase 3 Clinical Trial. Am J Respir Crit Care Med. 2021;203:585–93. doi:10.1164/rccm.202008-3177oc
- 30Kalydeco 25 mg Granules in Sachet. Electronic Medicines Compendium. 2021.https://www.medicines.org.uk/emc/product/10982/smpc (accessed Oct 2022).
- 31NHS England concludes wide-ranging deal for cystic fibrosis drugs. NHS England. 2019.https://www.england.nhs.uk/2019/10/nhs-england-concludes-wide-ranging-deal-for-cystic-fibrosis-drugs (accessed Oct 2022).
- 32Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508delCFTR. N Engl J Med. 2015;373:220–31. doi:10.1056/nejmoa1409547
- 33Milla CE, Ratjen F, Marigowda G, et al. Lumacaftor/Ivacaftor in Patients Aged 6–11 Years with Cystic Fibrosis and Homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017;195:912–20. doi:10.1164/rccm.201608-1754oc
- 34McNamara JJ, McColley SA, Marigowda G, et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. The Lancet Respiratory Medicine. 2019;7:325–35. doi:10.1016/s2213-2600(18)30460-0
- 35Long-term Safety of Lumacaftor/Ivacaftor in Subjects With Cystic Fibrosis Who Are Homozygous for F508del and 12 to <24 Months of Age at Treatment Initiation. ClinicalTrials.gov. 2020.https://clinicaltrials.gov/ct2/show/NCT04235140 (accessed Oct 2022).
- 36Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med. 2017;377:2013–23. doi:10.1056/nejmoa1709846
- 37Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med. 2017;377:2024–35. doi:10.1056/nejmoa1709847
- 38Munck A, Kerem E, Ellemunter H, et al. Tezacaftor/ivacaftor in people with cystic fibrosis heterozygous for minimal function CFTR mutations. Journal of Cystic Fibrosis. 2020;19:962–8. doi:10.1016/j.jcf.2020.04.015
- 39Walker S, Flume P, McNamara J, et al. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11 years with cystic fibrosis. Journal of Cystic Fibrosis. 2019;18:708–13. doi:10.1016/j.jcf.2019.06.009
- 40Davies JC, Sermet-Gaudelus I, Naehrlich L, et al. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. Journal of Cystic Fibrosis. 2021;20:68–77. doi:10.1016/j.jcf.2020.07.023
- 41Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. The Lancet. 2019;394:1940–8. doi:10.1016/s0140-6736(19)32597-8
- 42Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med. 2019;381:1809–19. doi:10.1056/nejmoa1908639
- 43Barry PJ, Mall MA, Álvarez A, et al. Triple Therapy for Cystic Fibrosis Phe508del–Gating and –Residual Function Genotypes. N Engl J Med. 2021;385:815–25. doi:10.1056/nejmoa2100665
- 44Zemanick ET, Taylor-Cousar JL, Davies J, et al. A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am J Respir Crit Care Med. 2021;203:1522–32. doi:10.1164/rccm.202102-0509oc
- 45Evaluation of ELX/TEZ/IVA in Cystic Fibrosis (CF) Subjects 2 Through 5 Years. ClinicalTrials.gov. 2020.https://clinicaltrials.gov/ct2/show/NCT04537793 (accessed Oct 2022).
- 46Landmark NHS deal to open up access to life-changing cystic fibrosis drug. NHS England. 2020.https://www.england.nhs.uk/2020/08/landmark-nhs-deal-to-open-up-access-to-life-changing-cystic-fibrosis-drug (accessed Oct 2022).
- 47‘Miracle’ Cystic Fibrosis treatment for children on the NHS. NHS England. 2022.https://www.england.nhs.uk/2022/01/miracle-cystic-fibrosis-treatment-for-children-on-the-nhs (accessed Oct 2022).
- 48Updated Commissioning Statement Ivacaftor, tezacaftor/ivacaftor, lumacaftor/ivacaftor and elexacaftor/tezacaftor/ivacaftor for licensed and off-label use in patients with cystic fibrosis who have named mutations. NHS England. 2022.https://www.england.nhs.uk/wp-content/uploads/2021/03/Commissioning-Statement-CF-modulator-therapies-for-Cystic-Fibrosis-UPDATED-2022.pdf?UNLID=776533918202237122216 (accessed Oct 2022).
- 49Specialised Services Policy Position Statement PP198. Cystic Fibrosis Modulator Therapies. Welsh Health Specialised Services Committee. 2022.https://whssc.nhs.wales/commissioning/whssc-policies/cystic-fibrosis/cystic-fibrosis-modulator-therapies-policy-position-statement-pp198-march-2022/ (accessed Oct 2022).
- 50Use of Kaftrio for Younger Cystic Fibrosis Patients Confirmed. Department of Health, Northern Ireland. 2022.https://www.health-ni.gov.uk/news/use-kaftrio-younger-cystic-fibrosis-patients-confirmed (accessed Oct 2022).
- 51Treating cystic fibrosis. NHS Scotland. 2020.https://www.gov.scot/news/treating-cystic-fibrosis/ (accessed Oct 2022).
- 52FDA Approves Expansion of Modulators for People With Certain Rare Mutations. Cystic Fibrosis Foundation. 2020.https://www.cff.org/news/2020-12/fda-approves-expansion-modulators-people-certain-rare-mutations (accessed Oct 2022).
- 53McGarry ME, McColley SA. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatric Pulmonology. 2021;56:1496–503. doi:10.1002/ppul.25285
- 54Desai M, Hine C, Whitehouse JL, et al. Who are the 10%? – Non eligibility of cystic fibrosis (CF) patients for highly effective modulator therapies. Respiratory Medicine. 2022;199:106878. doi:10.1016/j.rmed.2022.106878
- 55Konstan MW, VanDevanter DR, Rowe SM, et al. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF). Journal of Cystic Fibrosis. 2020;19:595–601. doi:10.1016/j.jcf.2020.01.007
- 56Drug development pipeline. Cystic Fibrosis Foundation. https://apps.cff.org/trials/pipeline (accessed Oct 2022).
- 57Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19:939–45. doi:10.1038/nm.3201
- 58Dekkers JF, Berkers G, Kruisselbrink E, et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016;8. doi:10.1126/scitranslmed.aad8278
- 59HIT-CF. HIT-CF Europe. https://www.hitcf.org/ (accessed Oct 2022).
- 60Sly PD, Brennan S, Gangell C, et al. Lung Disease at Diagnosis in Infants with Cystic Fibrosis Detected by Newborn Screening. Am J Respir Crit Care Med. 2009;180:146–52. doi:10.1164/rccm.200901-0069oc
- 61Dolgin E. In utero intervention to stem the damage of cystic fibrosis. Nature. 2020;583:S6–7. doi:10.1038/d41586-020-02108-8
- 62Duckers J, Lesher B, Thorat T, et al. Real-World Outcomes of Ivacaftor Treatment in People with Cystic Fibrosis: A Systematic Review. JCM. 2021;10:1527. doi:10.3390/jcm10071527
- 63Mitchell RM, Jones AM, Stocking K, et al. Longitudinal effects of ivacaftor and medicine possession ratio in people with the Gly551Asp mutation: a 5-year study. Thorax. 2021;76:874–9. doi:10.1136/thoraxjnl-2020-215556
- 64Volkova N, Moy K, Evans J, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. Journal of Cystic Fibrosis. 2020;19:68–79. doi:10.1016/j.jcf.2019.05.015
- 65Guimbellot JS, Baines A, Paynter A, et al. Long term clinical effectiveness of ivacaftor in people with the G551D CFTR mutation. Journal of Cystic Fibrosis. 2021;20:213–9. doi:10.1016/j.jcf.2020.11.008
- 66Barry PJ, Taylor-Cousar JL. Triple combination cystic fibrosis transmembrane conductance regulator modulator therapy in the real world – opportunities and challenges. Current Opinion in Pulmonary Medicine. 2021;27:554–66. doi:10.1097/mcp.0000000000000819
- 67A Prospective Study to Evaluate Biological and Clinical Effects of Significantly Corrected CFTR Function (PROMISE). ClinicalTrials.gov. 2019.https://clinicaltrials.gov/ct2/show/NCT04038047 (accessed Oct 2022).
- 68The PROMISE Pediatric Study 6 to 11 Years Old. ClinicalTrials.gov. 2020.https://clinicaltrials.gov/ct2/show/NCT04613128 (accessed Oct 2022).
- 69Real World Clinical Outcomes With Novel Modulator Therapy Combinations in People With CF (RECOVER). ClinicalTrials.gov. 2020.https://clinicaltrials.gov/ct2/show/NCT04602468 (accessed Oct 2022).
- 70Study to Evaluate Biological & Clinical Effects of Significantly Corrected CFTR Function in Infants & Young Children (BEGIN). ClinicalTrials.gov. 2020.https://clinicaltrials.gov/ct2/show/NCT04509050 (accessed Oct 2022).
- 71Dagenais RVE, Su VC, Quon BS. Correction: Dagenais et al. Real-World Safety of CFTR Modulators in the Treatment of Cystic Fibrosis: A Systematic Review. J. Clin. Med. 2021, 10, 23. JCM. 2022;11:318. doi:10.3390/jcm11020318
- 72Ivacaftor, tezacaftor, elexacaftor (Kaftrio) in combination with ivacaftor (Kalydeco): risk of serious liver injury; updated advice on liver function testing. Medicines and Healthcare Products Regulatory Agency. 2022.https://www.gov.uk/drug-safety-update/ivacaftor-tezacaftor-elexacaftor-kaftriov-in-combination-with-ivacaftor-kalydeco-risk-of-serious-liver-injury-updated-advice-on-liver-function-testing (accessed Oct 2022).
- 73Baiardini I, Steinhilber G, DI M, et al. Anxiety and depression in cystic fibrosis. Minerva Med 2015;106:1–8.https://www.ncbi.nlm.nih.gov/pubmed/27427260
- 74Sergeev V, Chou FY, Lam GY, et al. The Extrapulmonary Effects of Cystic Fibrosis Transmembrane Conductance Regulator Modulators in Cystic Fibrosis. Annals ATS. 2020;17:147–54. doi:10.1513/annalsats.201909-671cme
- 75McKinzie CJ, Goralski JL, Noah TL, et al. Worsening anxiety and depression after initiation of lumacaftor/ivacaftor combination therapy in adolescent females with cystic fibrosis. Journal of Cystic Fibrosis. 2017;16:525–7. doi:10.1016/j.jcf.2017.05.008
- 76Talwalkar JS, Koff JL, Lee HB, et al. Cystic Fibrosis Transmembrane Regulator Modulators: Implications for the Management of Depression and Anxiety in Cystic Fibrosis. Psychosomatics. 2017;58:343–54. doi:10.1016/j.psym.2017.04.001
- 77Heo S, Young DC, Safirstein J, et al. Mental status changes during elexacaftor/tezacaftor / ivacaftor therapy. Journal of Cystic Fibrosis. 2022;21:339–43. doi:10.1016/j.jcf.2021.10.002
- 78Barry PJ, Plant BJ, Nair A, et al. Effects of Ivacaftor in Patients With Cystic Fibrosis Who Carry the G551D Mutation and Have Severe Lung Disease. Chest. 2014;146:152–8. doi:10.1378/chest.13-2397
- 79Bailey J, Rozga M, McDonald CM, et al. Effect of CFTR Modulators on Anthropometric Parameters in Individuals with Cystic Fibrosis: An Evidence Analysis Center Systematic Review. Journal of the Academy of Nutrition and Dietetics. 2021;121:1364-1378.e2. doi:10.1016/j.jand.2020.03.014
- 80Collins S, Lowdon J, Watson K-L, et al. P166 “The party is over because I nowhave a sensible diet” – the experience of people with cystic fibrosis (CF) on CFTR modulators. Journal of Cystic Fibrosis. 2022;21:S112. doi:10.1016/s1569-1993(22)00497-0
- 81Rigon S, Brignole C, Paiola G, et al. P159 Modifications of anthropometric parameters and body composition after Kaftrio® in a group of adolescents and young adults. Journal of Cystic Fibrosis. 2022;21:S109–10. doi:10.1016/s1569-1993(22)00490-8
- 82Barrett J, Patel N, Rashid R, et al. P161 Body mass index change in adult patients with cystic fibrosis following the introduction of triple CFTR therapy elexacaftor/tezacaftor/ivacaftor: a regional adult cystic fibrosis centre experience. Journal of Cystic Fibrosis. 2022;21:S110. doi:10.1016/s1569-1993(22)00492-1
- 83Zamponi V, Cirilli N, Mazzoni N, et al. P162 Body composition assessment in cystic fibrosis (CF) patients on elexacaftor/texacaftor/ivacaftor. Journal of Cystic Fibrosis. 2022;21:S110–1. doi:10.1016/s1569-1993(22)00493-3
- 84Jain R, Taylor-Cousar JL. Fertility, Pregnancy and Lactation Considerations for Women with CF in the CFTR Modulator Era. JPM. 2021;11:418. doi:10.3390/jpm11050418
- 85Jain R, Kazmerski TM, Zuckerwise LC, et al. Pregnancy in cystic fibrosis: Review of the literature and expert recommendations. Journal of Cystic Fibrosis. 2022;21:387–95. doi:10.1016/j.jcf.2021.07.019
- 86Taylor-Cousar JL, Jain R. Maternal and fetal outcomes following elexacaftor-tezacaftor-ivacaftor use during pregnancy and lactation. Journal of Cystic Fibrosis. 2021;20:402–6. doi:10.1016/j.jcf.2021.03.006
- 87Taylor-Cousar JL. CFTR Modulators: Impact on Fertility, Pregnancy, and Lactation in Women with Cystic Fibrosis. JCM. 2020;9:2706. doi:10.3390/jcm9092706
- 88Nash EF, Middleton PG, Taylor-Cousar JL. Outcomes of pregnancy in women with cystic fibrosis (CF) taking CFTR modulators – an international survey. Journal of Cystic Fibrosis. 2020;19:521–6. doi:10.1016/j.jcf.2020.02.018
- 89Prospective Study of Pregnancy in Women With Cystic Fibrosis (MAYFLOWERS) . ClinicalTrials.gov. 2021.https://clinicaltrials.gov/ct2/show/NCT04828382 (accessed Oct 2022).
- 90Eakin MN, Bilderback A, Boyle MP, et al. Longitudinal association between medication adherence and lung health in people with cystic fibrosis. Journal of Cystic Fibrosis. 2011;10:258–64. doi:10.1016/j.jcf.2011.03.005
- 91Kirkham HS, Staskon F, Hira N, et al. Outcome evaluation of a pharmacy-based therapy management program for patients with cystic fibrosis. Pediatr Pulmonol. 2018;53:720–7. doi:10.1002/ppul.23978
- 92Siracusa CM, Ryan J, Burns L, et al. Electronic monitoring reveals highly variable adherence patterns in patients prescribed ivacaftor. Journal of Cystic Fibrosis. 2015;14:621–6. doi:10.1016/j.jcf.2015.05.009
- 93Olivereau L, Nave V, Garcia S, et al. Adherence to lumacaftor-ivacaftor therapy in patients with cystic fibrosis in France. Journal of Cystic Fibrosis. 2020;19:402–6. doi:10.1016/j.jcf.2019.09.018
- 94Mehta Z, Kamal KM, Miller R, et al. Adherence to cystic fibrosis transmembrane conductance regulator (CFTR) modulators: analysis of a national specialty pharmacy database. Journal of Drug Assessment. 2021;10:62–7. doi:10.1080/21556660.2021.1912352
- 95Marrero Álvarez P, Fernández Polo A, Cañete Ramírez C, et al. 4CPS-197 Evaluation of adherence to cystic fibrosis transmembrane conductance protein modulator drugs. Section 4: Clinical pharmacy services. 2022. doi:10.1136/ejhpharm-2022-eahp.198
- 96Howell I, Tugwell A, Bhaskaran D, et al. S59 Adherence to nebulised therapies in people with cystic fibrosis starting Elexacaftor/Tezacaftor/Ivacaftor (Kaftrio). Treatment choices in cystic fibrosis and bronchiectasis: what works and when. 2021. doi:10.1136/thorax-2021-btsabstracts.65
- 97Rowbotham NJ, Smith S, Leighton PA, et al. The top 10 research priorities in cystic fibrosis developed by a partnership between people with CF and healthcare providers. Thorax. 2017;73:388–90. doi:10.1136/thoraxjnl-2017-210473
- 98A registry-based trial investigating therapy rationalisation in people with cystic fibrosis (the CF STORM study). National Institute of Health Research. Applied Research Collaboration East of England. https://arc-eoe.nihr.ac.uk/research-implementation/research-themes/health-economics-and-prioritisation/registry-based-trial (accessed Oct 2022).
- 99Impact of Discontinuing Chronic Therapies in People With Cystic Fibrosis on Highly Effective CFTR Modulator Therapy (SIMPLIFY) . ClinicalTrials.gov. 2020.https://clinicaltrials.gov/ct2/show/NCT04378153 (accessed Oct 2022).
- 100Home-Reported Outcomes in People With Cystic Fibrosis Taking Highly Effective CFTR Modulator Therapy . ClinicalTrials.gov. 2021.https://clinicaltrials.gov/ct2/show/NCT04798014 (accessed Oct 2022).
- 101A Phase 2 Study to Evaluate Efficacy and Safety of VX-561 in Subjects Aged 18 Years and Older With Cystic Fibrosis. ClinicalTrials.gov. 2019.https://clinicaltrials.gov/ct2/show/NCT03911713 (accessed Oct 2022).
- 102Floto RA, Olivier KN, Saiman L, et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax. 2015;71:i1–22. doi:10.1136/thoraxjnl-2015-207360
- 103Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & Therapeutics. 2013;138:103–41. doi:10.1016/j.pharmthera.2012.12.007
- 104van der Meer R, Wilms EB, Sturm R, et al. Pharmacokinetic interactions between ivacaftor and cytochrome P450 3A4 inhibitors in people with cystic fibrosis and healthy controls. Journal of Cystic Fibrosis. 2021;20:e72–6. doi:10.1016/j.jcf.2021.04.005
- 105G S, L G, K P, et al. Dose adjustments of Elexacaftor/Tezacaftor/Ivacaftor in response to mental health side effects in adults with cystic fibrosis. Journal of Cystic Fibrosis. 2022. doi:10.1016/j.jcf.2022.05.001
- 106Bentley S, Castellani C, Peckham D, et al., editors. OPTIMIZING PHARMACEUTICAL CARE IN CYSTIC FIBROSIS. European Cystic Fibrosis Society 2020. http://react-profile.org/ebook/ECFS_Book_2020/files/assets/common/downloads/ECFS%202020%20-%20Optimizing%20pharmaceutical%20care%20in%20cystic%20fibrosis.pdf?uni=b57b27cdee7b81f3b7996ab58da431c5 (accessed Oct 2022).
- 107Shaw N, Collins S, Smith T, et al. Optimising the care and quality of life of people with cystic fibrosis: the influence of cystic fibrosis transmembrane conductance regulator modulators. Br J Hosp Med. 2021;82:1–6. doi:10.12968/hmed.2021.0530
- 108Rang C, Keating D, Wilson J, et al. Re-imagining cystic fibrosis care: next generation thinking. Eur Respir J. 2020;55:1902443. doi:10.1183/13993003.02443-2019
- 109Page A, Goldenberg A, Matthews AL. Lived experiences of individuals with cystic fibrosis on CFTR-modulators. BMC Pulm Med. 2022;22. doi:10.1186/s12890-022-01825-2
- 110Dancing, golfing, and hiking the Ochil hills: one year of Kaftrio. Cystic Fibrosis Trust. https://www.cysticfibrosis.org.uk/news/dancing-golfing-and-hiking-the-ochil-hills-one-year-of-kaftrio-0 (accessed Oct 2022).
- 111Kaftrio – complex and individual experiences. Cystic Fibrosis Trust. https://www.cysticfibrosis.org.uk/sites/default/files/2022-04/Kaftrio%20-%20complex%20and%20invidual%20experiences.pdf (accessed Oct 2022).
- 112Havermans T, Duff AJA. Changing landscape: psychological care in the era of cystic fibrosis transmembrane conductance regulator modulators. Current Opinion in Pulmonary Medicine. 2020;26:696–701. doi:10.1097/mcp.0000000000000727
- 113Mercier J, Ruffin M, Corvol H, et al. Gene Therapy: A Possible Alternative to CFTR Modulators? Front. Pharmacol. 2021;12. doi:10.3389/fphar.2021.648203
- 114Lee J-A, Cho A, Huang EN, et al. Gene therapy for cystic fibrosis: new tools for precision medicine. J Transl Med. 2021;19. doi:10.1186/s12967-021-03099-4
- 115Development of gene therapy treatment for cystic fibrosis moves to the next stage . Cystic Fibrosis Trust. https://www.cysticfibrosis.org.uk/news/development-of-gene-therapy-treatment-for-cystic-fibrosis-moves-to-the-next-stage (accessed Oct 2022).
- 116Study to Evaluate the Safety & Tolerability of MRT5005 Administered by Nebulization in Adults With Cystic Fibrosis (RESTORE-CF) . ClinicalTrials.gov. 2017.https://clinicaltrials.gov/ct2/show/NCT03375047 (accessed Oct 2022).
- 117Hodges CA, Conlon RA. Delivering on the promise of gene editing for cystic fibrosis. Genes & Diseases. 2019;6:97–108. doi:10.1016/j.gendis.2018.11.005
- 118Balfour-Lynn IM, King JA. CFTR modulator therapies – Effect on life expectancy in people with cystic fibrosis. Paediatric Respiratory Reviews. 2022;42:3–8. doi:10.1016/j.prrv.2020.05.002
- 119Parkins MD, Parkins VM, Rendall JC, et al. Changing epidemiology and clinical issues arising in an ageing cystic fibrosis population. Therapeutic Advances in Respiratory. 2010;5:105–19. doi:10.1177/1753465810386051
- 120Keogh RH, Tanner K, Simmonds NJ, et al. The changing demography of the cystic fibrosis population: forecasting future numbers of adults in the UK. Sci Rep. 2020;10. doi:10.1038/s41598-020-67353-3
- 121Hadjiliadis D, Khoruts A, Zauber AG, et al. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations. Gastroenterology. 2018;154:736-745.e14. doi:10.1053/j.gastro.2017.12.012