Abstract
Aim
To determine the microbiological relevance of syringe hub fluid to product integrity during storage.
Design
Aseptic preparation of 1,002 luer-lock syringes under European Community Good Manufacturing Practice (EC GMP) Grade A conditions with (500) and without (502) a hub fluid reservoir subsequently stored for 28 days in an uncontrolled environment followed by a determination of microbiological contamination of each syringe hub and fill. Positive and negative controls were used to ensure the specificity and selectivity of results.
Outcome measures
Level of microbiological contamination of the syringe hub and syringe fill.
Results
The 500 syringes with hub fluid showed microbiological contamination in 2 hubs and no fills. The 502 syringes without hub fluid showed microbiological contamination in 4 hubs and no fills. No statistically significant difference in contamination of syringe hubs (P=0.687) or syringe fills (P>0.05) was detected but a statistically significant difference between syringe hubs and syringe fills was found (P=0.031).
Conclusions
This study demonstrated that the presence of hub fluid did not increase microbiological contamination of syringes stored for up to 28 days. The microbiological contamination of syringe hubs appears greater than syringe fills after a storage period of 28 days (P=0.031).
The parenteral administration of the contents of a contaminated syringe presents a real risk to a patient, particularly since micro-organisms that normally present no pathological risk may do so when given parenterally.1 Contaminated products have led to fatalities2 and up to a third of nosocomial bacteraemia occurs in association with intravenous infusions and catheters.3 A drug giving port can become contaminated by the use of a contaminated product4–6 and this contamination can subsequently be administered to the patient when the same port is used again.5 Any contamination of a syringe hub is therefore a potential risk to a patient because it may contaminate the giving port when the syringe contents are administered.
The fitness for purpose (and therefore quality) of an aseptically prepared product can be measured against various criteria, but one of the most important is the absence of microbiological contamination.7 A European Community Good Manufacturing Practice (EC GMP) Grade A environment controls the background levels of viable, particulate and chemical contamination and therefore reduces the probability of any such contamination being incorporated into the final product during aseptic preparation. The limits for particles and micro-organisms within an EC GMP Grade A environment are controlled by the Standard BS EN ISO 14644-1: 19998 and by EC GMP requirements.9,10 Although these limits must be complied with for aseptic preparation of parenteral products that may be stored before administration to a patient, it is not necessary for them to be adhered to in the aseptic preparation of products for immediate administration to a patient, such as takes place on a hospital ward.
Products for immediate parenteral use prepared aseptically in a clinical environment have a relatively high probability of microbiological contamination (0.02 to 0.254,11) compared with products prepared aseptically under EC GMP Grade A conditions (0.00001 to 0.00212,13). However, products prepared under EC GMP Grade A conditions may be stored before administration to a patient, and it is during this storage period that the products can be at the greatest risk from both microbiological contamination and the subsequent growth of any micro-organisms present.
During aseptic preparation a fluid reservoir may unintentionally be created in the hub (luer-thread14) of a syringe, which may compromise the product integrity during storage by potentially providing a specific opportunity for microbiological contamination to gain access to the syringe hub and subsequently the syringe fill (Figure 1).

Following aseptic preparation, a “sterility test”15 may be carried out and, in combination with quality assurance processes, can confirm the absence of microbiological contamination of prepared products. A hub fluid reservoir may have implications for the fitness for intended purpose of a final product because the growth of micro-organisms can be rapid1 and therefore any potential contamination reservoirs in syringes stored before administration may present a considerable infection risk since no microbiological testing of syringes is carried out immediately before administration.
Previous studies of the risk of external microbiological contamination accessing the internal aspects of a syringe during storage have focused on the potential for microbiological contamination to access the syringe fill via the plunger-barrel interface,3,16,17 or have not considered how external contamination during storage may access the syringe.18 Poor study design, including low study power and the use of syringe fills with inherent antimicrobial activity also limits the value of previous studies.18
Microbial growth requires the presence of available water, different microbes varying in their tolerance to low available water levels.19
This indicates that the survival and growth of micro-organisms within a syringe hub may require a minimum level of water, and a dry hub may not allow proliferation of microbiological contamination. The various fluids used for syringe fills may have an impact on microbiological contamination and growth. For example, it has been shown that 5 per cent w/v glucose is more likely to support microbiological contamination than 0.9 per cent w/v sodium chloride.20 The potential for microbiological contamination is not limited to diluents, with examples of intravenous nutrition emulsions and propofol having been shown as suitable growth mediums for microorganisms.21–23 There may also be a possibility of prepared cytotoxic doses being able to support the growth of micro-organisms.2,24 Following from this, the most likely fluid to support the growth of common environmental contaminants would be a medium specifically designed for this purpose, such as tryptone soy broth. Tryptone soy broth was therefore selected as the liquid medium of choice for the practical aspect of this research in order to provide a high level of challenge to the stored syringes.
The principal aim of this research was to indicate the suitability of syringes with hub fluid for final release. Only luer-lock syringes were considered because luer-slip designs should not allow the collection of fluid in the same way because they have no screw-thread at the syringe hub.
We aimed to find out whether:
- Syringe hub fluid allows microbiological contamination to access and proliferate within the hub itself during a storage period of 28 days
- Syringe hub contamination can subsequently access and proliferate within the syringe fill during a storage period of 28 days
- The microbiological contamination of the syringe hub is greater than the syringe fill after a storage period of 28 days
Method
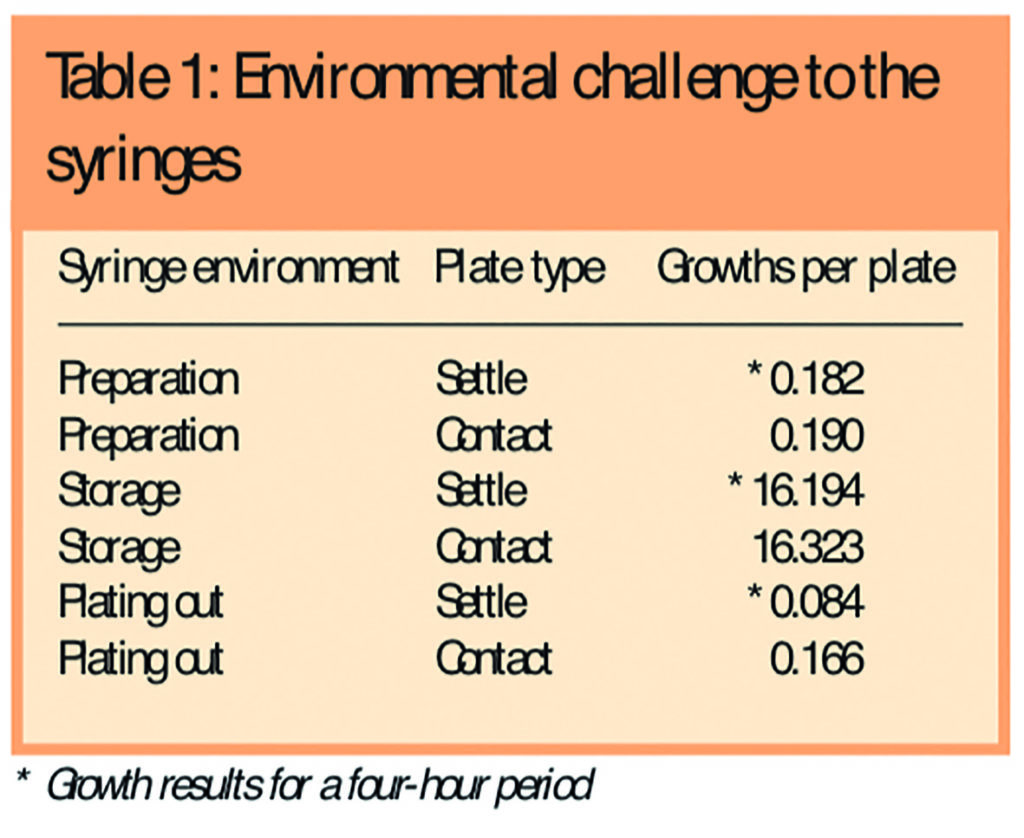
A total of 1,002 10ml luer-lock syringes (Beckton-Dickenson, Oxford, UK) were prepared with a 2ml fill of single strength tryptone soy broth (Torbay Pharmacy ManufacturingUnit,Torbay,UK) in batches of up to 108 syringes under EC GMP grade A conditions using strict aseptic technique. Each syringe was capped with a Combi-cap (Universal Hospital Supplies Ltd, London, UK). The prepared syringes were divided into those with hub fluid and those without hub fluid by a single pharmacist. Segregation between these two groups was maintained throughout the remaining study period. All syringes were stored for 28 days at room temperature (23.3C; range 21.2C–25.5C) in an uncontrolled environment where the microbiological challenge to the syringes was regularly confirmed with both four-hour settle plates and contact plates (used under the stored syringes, followed by the use of an alcohol wipe to remove any residual agar).
Following the 28-day storage period, syringes were transferred back into an EC GMP Grade A environment taking care to avoid spraying the 70 per cent w/v alcohol directly at the syringe hub. Each syringe cap was removed and the tip of the syringe along with all internal surfaces of the cap were swabbed using a pre-moistened sterile swab (Medical Wire & Co, Bath, UK) which was subsequently plated out onto an agar plate. Care was taken to swab only internal surfaces and to avoid any contact between the swab and any surfaces of the prepared syringe that had been exposed to any uncontrolled environment. A new cap was applied to the syringe, which was subsequently incubated for 14 days at 32C (±0.5C) to identify any contamination of the syringe fill. The swab plates were incubated for seven days at 32C (±0.5C) to represent any contamination of the syringe hub. The incubation temperature used was to maximise the recovery of any bacteria or fungi present. Following the appropriate incubation period, syringes were examined for turbidity and plates were examined for growth quantitatively.
Negative controls were provided by incubating 10 syringes without hub fluid along with the remainder of the tryptone soy broth in the original bottles from each batch for 14 days at 32C (±0.5C). With no growth (defined as no visible turbidity) these syringes were subsequently stored for four further weeks at room temperature (23.3C; range 21.2C–25.5C) in the same uncontrolled environment as the main syringe groups. Results from the negative control syringes were included in the final results.
A positive control was provided by drawing up a 2ml fill of single strength tryptone soy broth containing approximately 1x106cfu/ml of erythromycin-resistant Staphylococcus aureus into 10 10ml syringes. These syringes were incubated at 30C (range 24C–32C) for 14 days. To demonstrate recovery after storage, a one-drop sample was taken from the same syringe each time and plated out onto agar plates on days 2, 4, 7 and 12, which were subsequently incubated at 30C (range 24C–32C) for seven days.
Using SamplePower 2.0 and assuming a baseline contamination rate of 1 in 500 (or 0.002), for a Fisher Exact test it was determined that 500 syringes per group would give a power of approximately 80 per cent with a P≤0.05 detecting a 10 times relative risk difference between groups.
Results
Both the negative and positive controls confirmed the validity of the methods used and the subsequent results obtained with 100 per cent sensitivity and 100 per cent specificity. The preparation and plating out environments were shown to be within recommended limits for an EC GMP Grade A environment and the storage environment showed a significant microbiological challenge to the stored syringes (Table 1). No turbidity in any syringe was present immediately before swabbing, plating out and recapping.
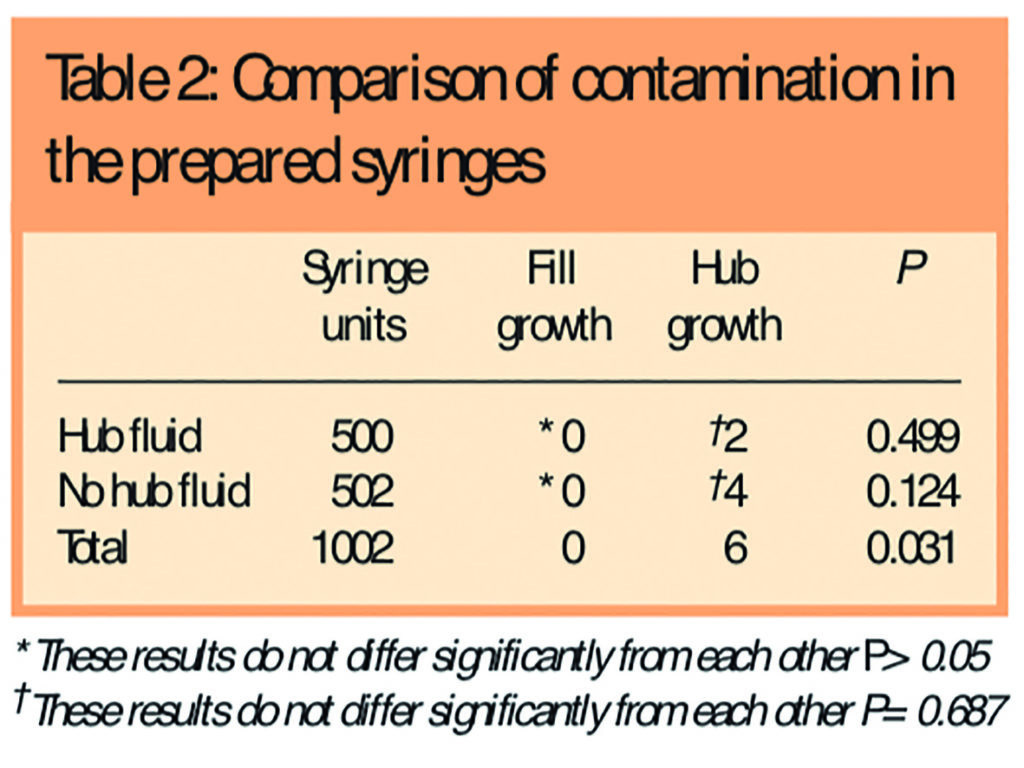
Five hundred syringes with hub fluid and 502 syringes without hub fluid were included. No syringe fill showed contamination. Two syringes with hub fluid and four syringes without hub fluid showed microbiological contamination of the hubs with common environmental organisms (Staphylococcus spp or Bacillus spp). Independent identification of contaminating micro-organisms was carried out using morphology.
Using a Fisher Exact test there was no statistically significant difference in microbiological contamination between the hubs (P=0.687) or the fills (P>0.05) of the two groups. The two groups can therefore be considered together as one larger group to compare syringe fill against syringe hub contamination. The level of microbiological contamination found in the syringe hubs was statistically greater than that found in the syringe fills (P=0.031). Neither of the two individual groups achieve statistical significance on their own due to insufficient numbers (type 2 statistical error). The contamination of the syringes results are shown in Table 2. A comparison between the hub contamination rate of six in 1,002 found in this study against suggested fill contamination rates for products prepared under controlled conditions12 gives a P<0.01 using a Fisher Exact test.
Discussion
If the prepared syringes were free from microbiological contamination on preparation, for any microbiological contamination to become present the syringe contents it would first have to contaminate the syringe hub or the plunger seal. Under the conditions of the study, it is apparent that microbiological contamination may be able get into the syringe hub but not the syringe fill over a storage period of 28 days. For syringes prepared within EC GMP Grade A conditions, the rate of microbiological contamination of the syringe hubs (six in 1,002) statistically exceeds published figures for the contamination rate of syringe fills.
Although the presence of hub fluid does not appear to affect the microbiological contamination rate of syringes stored for 28 days, there does appear to be an inherent risk of microbiological syringe hub contamination. This raises the possibility that the greatest microbiological risk to luer-lock syringe integrity is at the hub. The recovery rate from the syringe cap and hub using swabbing may have been less than 100 per cent and, therefore the contamination rate indicated may be lower than that which actually exists.
The preparation of parenteral doses within a controlled EC GMP Grade A environment with dedicated staff has numerous advantages,25–27 and is often recommended.28,29 The probability of a contaminated dose unit is reduced21 and the availability of pre-filled syringes with prolonged expiry dates improves patient safety and reduces staff stress levels.25 Patient safety is not based solely on microbiological risk, but also on other factors such as the often complex calculations required to prepare a dose.30
Broth tests are used to validate operators (and processes) and involve the aseptic manipulation of a broth medium which readily supports microbiological growth.31,32 In practice, these may be carried out within uncontrolled or controlled environmental conditions. The prepared broth units are incubated and no growth in any unit along with verification of operator technique by a supervising senior member of staff are typically used to verify the ability of an operator to perform aseptic manipulations competently before they prepare doses intended for administration. Current national recommendations31 and the proposed “Universal operator broth transfer validation”33 consider microbiological contamination of the syringe fill only.
It is important to consider products as a whole, not just any particular aspect.34 The current practice of determining the microbiological contamination rate of syringes by growth within the syringe fill, both within validation studies and broth tests may, therefore, present a risk lower that that which actually exists.
The possibility that the hub contamination within this study was introduced during syringe preparation is unlikely because of the consistent use of strict aseptic technique when preparing and subsequently manipulating the syringes. However, no specific validation of the absence of hub contamination during syringe preparation was undertaken and therefore the possibility that hubs may have been contaminated during preparation must be considered for both the syringes prepared within this study and all aseptically prepared syringes. Specific testing for syringe hub contamination may therefore be warranted during operator and process validation for the aseptic preparation of syringes intended for parenteral administration.
The rejection of prepared syringes with hub fluid because of the potentially higher microbiological contamination risk of the product during storage compared with syringes without hub fluid may not be necessary and therefore a policy change in some aseptic units may be appropriate. Other considerations, such as acceptability of the appearance of the syringe and the potential for user exposure to any hub fluid, may still mean that the presence of syringe hub fluid is undesirable or unacceptable.
Conclusions
- Syringe hub fluid does not appear to allow microbiological contamination to access and proliferate within the hub itself during a 28-day storage period
- The microbiological contamination of the syringe hub appears greater than the syringe fill after a 28-day storage period
- Syringe hub contamination does not appear to access and proliferate within the syringe fill during a 28-day storage period
Acknowledgements
This research was carried out by Mr Austin in part completion of the Leeds University pharmaceutical technology and quality assurance MSc, which is directed by Tim Sizer. Thanks to Marinos Elia and Steve Wootton from the Institute of Human Nutrition, Southampton General Hospital for their assistance with the statistics and preparation of the paper.
This paper was accepted for publication on 1 December 2005.
About the authors
Peter Austin, MSc, MRPharmS, is senior pharmacist and Stephen Dixson, BPharm, MRPharmS, is principal pharmacist at Southampton General Hospital.
Correspondence to: Mr Austin at Pharmacy Department, Southampton General Hospital, Tremona Road, Southampton SO16 6YD (e-mail Peter.Austin@suht.swest.nhs.uk)
References
- Sharp J. Quality in the manufacture of medicines and other healthcare products. London:Pharmaceutical Press; 2000. p339.
- Farwell J. Aseptic dispensing for NHS patients. London: Department of Health; 1995.
- Olivier L, Kendoff D, Wolfhard U, Nast-Kolb D, Yazici M, Esche H. Modified syringe designprevents plunger-related contamination and flow-rate tests. Journal of Hospital Infection2003;53:140–3.
- Worthington T, Tebbs S, Moss H, Bevan V, Kilburn J, Elliott T. Are contaminated flush solutions anoverlooked source for catheter-related sepsis? Journal of Hospital Infection 2001;49:81–83.
- Dutta A, Malhotra S.Syringe contamination by propofol: a possible mechanism. CanadianJournal of Anesthesia 2001;48:714–5.
- Skidmore G. Reuse of syringes in anaesthesia practice. Canadian Journal of Anaesthesia1996;43:418.
- Sharp J. Quality in the manufacture of medicines and other healthcare products. London:Pharmaceutical Press; 2000. p26.
- British Standards Institution. Cleanrooms and associated controlled environments — part 1:classification of air cleanliness (BS EN ISO 14644-1). London: BSI; 1999.
- Medicines Control Agency. Rules and guidance for pharmaceutical manufacturers anddistributors. London: The Stationery Office; 2002. pp97–102.
- Beaney A. Quality assurance of aseptic preparation services (4th ed). London: PharmaceuticalPress; 2005. pp48–50.
- NHS North West Region Aseptic Dispensing Working Party. Maintaining Asepsis During thePreparation of Pharmaceutical Products. Manchester: NHS North West Region; 1997. p 20.
- Sharp J. Quality in the manufacture of medicines and other healthcare products. London:Pharmaceutical Press; 2000. pp333–9.
- Needle R. Clinical governance in aseptic compounding. Hospital Pharmacist 2000;1:192.
- British Standards Institution. Conical fittings with a 6% (Luer) taper for syringes, needles andcertain other medical equipment — lock fittings (BS EN 1707). London: BSI; 1997.
- British Pharmacopoeia Commission. British Pharmacopoeia (volume 2, appendix XVIA).London: The Stationery Office; 2001. ppA298–A303.
- Huey W, Newton D, Augustine S, Vejraska B, Mitrano F. Microbial contamination potential ofsterile disposable plastic syringes. American Journal of Hospital Pharmacy 1985;42:102–5.
- Mitrano F, Baptista R, Newton D, Augustine S. Microbial contamination potential of solutions inprefilled disposable syringes used with a syringe pump. American Journal of Hospital Pharmacy 1986;43:78–80.
- Driver R, Snyder I, North F, Fife T. Sterility of anesthetic and resuscitative drug syringes used in the obstetric operating room. Anesthesia and Analgesia 1998;86:994–7.
- Prescott L, Harley J, Klein D. Microbiology (2nd ed). Oxford: Wm C Brown Communications Inc; 1993. pp122–6.
- Farrington M, McGinnes J, Matthews I, Park G. Do infusions of midazolam and propofol pose an infection risk to critically ill patients? British Journal of Anaesthesia 1994;72:415–7.
- Langford S. Mortality and morbidity from the in-use microbial contamination of intravenous products. Hospital Pharmacist 1999;6:104–9.
- Allwood M. Microbiological risks in parenteral nutrition compounding. Nutrition 1997;13: 60–1.
- Langford S. Microbial survival in infusion fluids — the relevance to the management of aseptic facilities. Hospital Pharmacist 2000;7:228–36.
- Allwood M, Stanley A, Wright P. The cytotoxics handbook (4th ed) Oxford: Radcliffe Medical Press; 2002. p257.
- Webster C, Merry A, Ducat C. Safety, cost and predrawn emergency drugs. Anaesthesia 2001;56:818–20.
- Scurr C. Contamination of injections. British Medical Journal 1979;2:1005.
- Needle R, Sizer T. The CIVAS handbook — the centralised intravenous additive services reference. London: Pharmaceutical Press; 1998. p1.
- Breckenridge A. 1976 Report of the working party on the addition of drugs to intravenous infusion fluids (HC[76]9). London: The Commission; 1976.
- Audit Commission. A spoonful of sugar: medicines management in NHS hospitals. London: The Commission; 2001.
- The future of CIVAS — can the risks be managed? Hospital Pharmacist 2000;1:191–3.
- Beaney A. Quality assurance of aseptic preparation services (4th ed) London: Pharmaceutical Press; 2005. pp92–5.
- Needle R, Sizer T. The CIVAS handbook — the centralised intravenous additive services reference. London: Pharmaceutical Press; 1998. p10.
- National CIVAS Group – Working Party on Evidence-Based Justification of CIVA Services. Draft proposal for a universal operator broth transfer validation (draft 7). 2001. Available at: http://home.btclick.com/civas/universalbroth/universal-operator-brothtest.pdf (accessed 1 December 2005).
- Dean D. The Institute of Packaging, packaging of pharmaceuticals, packages and closures. Norwich: Whitefriars Press Ltd; 1983. p1.