Key points:
- The translational research to create the clinical strategy for long-term adjuvant anti-hormone therapy for oestrogen receptor (ER) positive breast cancer, has saved thousands of women’s lives following a diagnosis of early breast cancer.
- Extending adjuvant tamoxifen therapy for ten years following breast cancer surgery, results in dramatic decreases in mortality during the ten years after stopping tamoxifen; indicating that ‘longer is better’.
- Aromatase inhibitors used as long-term adjuvant therapy for post-menopausal ER-positive breast cancer patients, decrease side effects relative to tamoxifen (e.g. endometrial cancer, gynaecological concerns, and thrombotic events) however, bone density decreases occur.
- A strategy of a gonadotropin-releasing hormone super-agonist plus an aromatase inhibitor is an optimal adjuvant therapy in younger premenopausal breast cancer patients with ER-positive disease.
- Research to find an orally active Selective ER Downregulator (SERD) to replace the depot injectable fulvestrant, may be enhanced by finding “pseudo-SERDs” that destroy tumour ERs but improve women’s bone and heart health similar to Selective ER Modulator (SERM).
Introduction
Source: K H Fund / Science Photo Library
Coloured 3D magnetic resonance imaging (MRI) scan of a section through the breast of a female patient, showing a malignant tumour (blue). The appearance of the nipple (right), is distorted because of the presence of the tumour.
Breast cancer occurs in both women and men, and incidence increases with age[1]
. Breast cancer has the highest incidence in women, with 53,400 cases diagnosed in women in the UK in 2013[1]
. Male breast cancer has a much lower incidence, with 340 cases diagnosed in the UK in 2013[1]
. Around 30 years ago, breast cancer was the leading cause of death in women; however, this has since been surpassed by lung cancer[1]
. There are two reasons for this: lung cancer mortality increased because more women have smoked cigarettes, while breast cancer mortality decreased because of advances in screening and treatment, both of which significantly lowered death rates over the past two decades[2],
[3],
[4]
. The main advance in the treatment of breast cancer is the application of anti-oestrogenic treatment strategies that evolved slowly throughout the 20th century[5],
[6]
.
Based on individual observational clinical reports during the first half of the 20th century[5]
, the advance in breast cancer treatment began with endocrine ablation (ovary, adrenal, pituitary gland), which was subsequently found to regulate oestrogen synthesis. Endocrine ablation became the standard of care for the treatment of premenopausal or postmenopausal women with metastatic breast cancer (MBC), as did the paradoxical introduction of high-dose oestrogen therapy used for the treatment of postmenopausal MBC patients[7]
. Response rates to any treatment remained at only 30%[7]
. The question then became, in the second half of the 20th century; could a predictive test be devised to identify those patients who would be responsive to endocrine ablation?
The primary reason for this clinical test was to reduce morbidity and hospital stays for the majority of patients that would be unresponsive to endocrine therapy. The discovery of selective oestrogen binding in the oestrogen target tissues (uterus, vagina, pituitary gland) in animals was an important first step in understanding oestrogen action[8],
[9]
. The technology to detect the oestrogen receptor (ER) in breast tumours was advanced from the laboratory to clinical testing[10],
[11]
. It was reasoned that the ER would be present in those breast tumours that responded to endocrine ablation or high-dose oestrogen therapy (there were no clinical studies of tamoxifen in MBC related to tumour ERs at this time). Clinical studies in MBC confirmed that the absence of the ER in breast tumours was enough to identify patients with tumours unlikely to respond to endocrine treatment[12]
. By contrast, patients with ER-positive tumours had a high likelihood of a response to any endocrine therapy[12]
. Although a predictive test to identify those patients who would be spared endocrine therapy if their tumours were ER-negative was a significant advance in patient care, it was the realisation that the ER could be used as a target for an anti-oestrogenic drug (tamoxifen) that became the important advance[6],
[13],
[14]
. A recent analysis of the important advances in medical oncology during the past 50 years concluded that “the development of therapeutics for ER-expressing breast cancers has been one of the great clinical advances of the past 50 years and has served as a paradigm for the development of targeted therapies in oncology”, and, “as most breast cancers are ER–positive, and given the worldwide prevalence of the diseases, it is arguable that anti-oestrogen treatments have had greater global impact than any other treatment intervention in cancer medicine”[15]
.
ERs located in the oestrogen target tissues (see ‘Figure 1: The hypothalamus pituitary-gonadal axis’), including the majority of breast cancers, of women, act as the signal transduction pathway for gene expression and cell replication at that target site. The non-steroidal anti-oestrogen tamoxifen and its metabolites are classified as competitive inhibitors of oestrogen binding to the ER[16]
, an action that blocks breast cancer tumour cell replication during treatment. Although tamoxifen is used for the treatment of all stages of breast cancer, primary prevention of breast cancer in high-risk populations, ductal carcinoma in situ (DCIS), and male breast cancer, the major strategic impact of tamoxifen in cancer therapeutics is long-term adjuvant therapy (five years) for patients that are stage I and stage II with a primary tumour that is ER-positive. The revolutionary and counterintuitive strategy to deploy long-term ‘anti-oestrogenic’ adjuvant therapy was a paradigm-changing approach to cancer treatment[6]
. The strategy conceived in the laboratory in the 1970s[6]
was counterintuitive because tamoxifen is only effective for a year or two in the treatment of MBC at the end of life, because in MBC, acquired drug resistance to tamoxifen develops rapidly with a large tumour bulk and, as a result, there is greater mutational variability. The clinical reasoning in the 1970s, when adjuvant trials with tamoxifen were started, was that only one or two years of tamoxifen treatment should be given because longer therapy (five years or forever) would result in early drug resistance[6]
. However, a low burden of micrometastases around a patient’s body clearly responds differently because longer treatment was subsequently shown to be superior to shorter adjuvant treatment in clinical trials[17],
[18]
. This successful translational research strategy is credited to have extended the lives of perhaps millions of breast cancer patients worldwide[6]
.
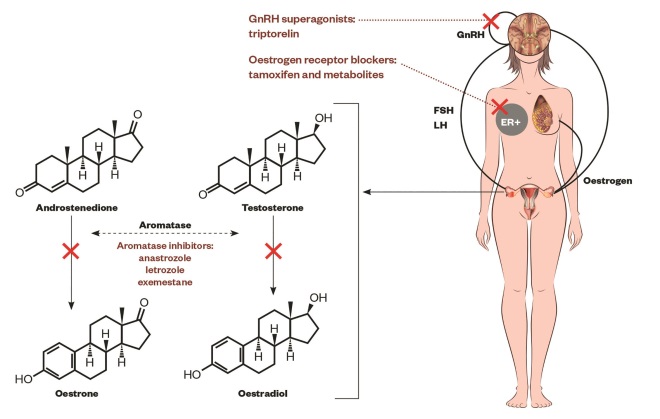
Figure 1: The hypothalamus pituitary-gonadal axis
Schematic representation of the hypothalamus pituitary-gonadal axis, the aromatisation of androgens (androstenedione, testosterone) into oestrogens (oestrone, oestradiol) by aromatase enzyme in granulosa cells in ovaries, oestrogen action on the breast promoting ER-positive cells and the sites of action of drugs[51],
[52],
[103],
[104]
: gonadotropin-releasing hormone (GnRH) superagonists (i.e. triptorelin or goserelin) alone, or with oestrogen receptor blockers (i.e. tamoxifen and metabolites) and aromatase inhibitors (i.e. anastrozole, letrozole, exemestane). GnRH-expressing neurons in the hypothalamus secretes GnRH. The anterior portion of the pituitary gland (adenohypophysis or pars anterior) produces luteinizing hormone (LH) and follicle-stimulating hormone (FSH), consequently the gonads (ovaries) produce oestrogen which in turn promotes the physiologic or pathologic growth of the breast.
The evolving understanding of the biology of breast cancer that occurred during the 20th century ultimately permitted the rational design of placebo-controlled, randomised clinical trials, correctly powered to identify the value of a therapeutic strategy compared with no therapy. This clinical advance was the most important step taken during the 1960s in clinical oncology and converted clinical practice from anecdotes, articulated by eminent physicians or surgeons based on their personal experiences, into evidence-based clinical practice as a universal standard of care.
The conversation between the laboratory and the clinic is a two-way process that has been enhanced by an understanding of laboratory models of acquired resistance to anti-hormonal therapies[19],
[20],
[21],
[22]
and the dissection of specific mechanisms of cellular adaptation and the plasticity of cell populations in the laboratory that mimic acquired resistance of breast cancer noted in clinical practice[23],
[24]
. Most importantly, an understanding of the molecular mechanisms of ER mutations that have been studied in the laboratory over the past 20 years have now been combined with the recent findings of mutations in the ER in the metastases of patients who failed aromatase inhibitor (AI) therapy[25]
. These data illustrate resistance mechanisms to AIs and selective ER modulators (SERMs)[25]
that have sparked a renewed enthusiasm to find new ‘pure anti-oestrogens’ (e.g. selective ER downregulators, or SERDs) that could be deployed within the next decade as new anti-hormone therapies in the adjuvant setting.
Sources and selection criteria
The identification of scientific studies referred to in this article are based on a library of references accumulated over the 45 years spanning the research that created the concepts and clinical principles described here. This is a personal account of what occurred, the results, and what is needed for the next advance in healthcare.
Aromatase inhibitors as an alternative to adjuvant tamoxifen treatment in postmenopausal patients
The plan to develop a specific medicine to target oestrogen synthesis in postmenopausal patients started in the early 1970s[26]
. Before that, and before tamoxifen was developed, several compounds were used clinically as endocrine agents of last resort. However, these medicines were not specific and affected multiple enzyme systems involved in the synthesis of other steroids, most notably, the glucocorticoids[26]
. The growing interest in tamoxifen as the first targeted agent in breast cancer therapy provided a new opportunity to develop a novel medicine without the oestrogen-like effect of tamoxifen. The theory was simply “there is no anti-oestrogen like no oestrogen at all”[27]
.
The targeted inhibition of the aromatase enzyme system CYP19 was first achieved with 4-hydroxyandrostenedione, an irreversible blocker of the enzyme system that aromatises androstenedione or testosterone to oestrogens (see figure 1). These pioneering studies in translational research[28],
[29]
set the stage for a pivotal ‘proof of principle’ clinical trial in MBC[30],
[31]
. However, 4-hydroxyandrostenedione was injectable and would ultimately have needed to compete against oral tamoxifen in an adjuvant therapy clinical trial. Nevertheless, the lone dominance of tamoxifen as the adjuvant endocrine therapy of choice from the mid-1980s[32]
not only created a valuable new targeted therapy with few serious side effects compared with cytotoxic chemotherapy, but also a major new market, as there was, as yet, no restriction on the duration of adjuvant tamoxifen therapy. The pharmaceutical industry responded to this opportunity with successive generations of orally-active aromatase inhibitors until the appropriate specific agents were found to block CYP19 with few side effects[14]
.
Today, there are three approved compounds: letrozole, anastrozole and exemestane. Letrozole and anastrozole are competitive inhibitors at the active site of the aromatase enzyme system, whereas exemestane is an irreversible blocker (i.e. a suicide inhibitor of the enzyme active site)[14]
.
At the same time, the clinically successful AIs were being rigorously investigated in MBC, a change occurred in the standard of care for the adjuvant therapy of breast cancer with tamoxifen. The National Surgical Adjuvant Breast and Bowel Project (NSABP) tested whether tamoxifen should be given for longer than five years in the adjuvant setting with an extension of the original NSABP B-14 placebo-controlled trial with node-negative ER-positive breast cancer patients. They concluded that “the benefits from five years of tamoxifen therapy persist through ten years of follow-up. No additional advantage is obtained from continuing tamoxifen therapy for more than five years”[33]
. Five years duration became the standard of care for anti-oestrogenic adjuvant treatment of ER-positive breast cancer. As a result, this duration became the benchmark for AIs to beat adjuvant tamoxifen treatment.
The first major adjuvant study, Arimidex, Tamoxifen Alone or in Combination (ATAC)[34]
, was designed to be a highly powered, stand-alone trial, to answer the question of whether anastrozole is superior to tamoxifen and whether the combination adjuvant treatment with tamoxifen and anastrozole for five years was superior to either individual agent. Each arm of the trial recruited more than 4,000 postmenopausal early breast cancer patients following surgery, with 7,839 (84%) tumours confirmed as ER-positive. Anastrozole was found to be superior to tamoxifen in the first analysis[34]
and subsequent reanalyses[35],
[36],
[37]
. Anastrozole was superior to either tamoxifen alone or the combination of tamoxifen and anastrozole. The fact that the combination of tamoxifen and anastrozole was essentially the same as tamoxifen at controlling disease reoccurrence, and with the same side effects, is worthy of explanation and comment.
It is a rule in pharmacology that if a full agonist (e.g. oestrogen) is completely removed from a receptor-mediated biological system (e.g. breast cancer), then the final response of the biological system will be quiescence with no receptor activation. Similarly, when a partial agonist/antagonist (e.g. tamoxifen) is introduced into the same receptor-mediated biological system (breast cancer), to block the action of a full agonist (oestrogen), then the final result of the system will reflect the intrinsic efficacy of the partial agonist/antagonist receptor complex (tamoxifen). In the ATAC trial, the anastrozole-treated arm removed all the full agonists (oestrone and oestradiol) to prevent growth of breast cancer through the ER signal transduction pathway. The addition of tamoxifen in this situation would be predicted to allow tamoxifen and its metabolites to bind to the breast tumour ER, resulting in the increased intrinsic efficacy of the ER complex. The weak oestrogen-like properties of tamoxifen would be predicted to enhance an increase in breast tumour cell replication. This action of tamoxifen in the presence of anastrozole in an oestrogen-deprived situation would result in a breast cancer recurrence rate the same as tamoxifen alone in the presence of basal postmenopausal oestrogen levels that are being blocked at the ER. This was demonstrated in the ATAC trial[34]
as the combination of anastrozole plus tamoxifen versus tamoxifen alone[36]
had the same anti-tumour properties: worse than anastrozole or “no oestrogen at all”[27]
.
The whole spectrum of adjuvant clinical trials comparing different AIs with tamoxifen are listed and reviewed in detail elsewhere[38],
[39]
and the scientific and strategic rationale for each of the AI trial designs are described in ‘Figure 2: The evolution of adjuvant trial design’. The designs of the adjuvant trials of five years of tamoxifen versus various AIs are: A: Anastrozole or letrozole alone for five years[36],
[40]
; B: Two to three years of tamoxifen followed by an AI for two to three years, or the reverse[40],
[41]
; C: Five years of adjuvant tamoxifen followed by five years of letrozole[42],
[43]
; and each of these can be compared with D: Five versus ten years of tamoxifen[18],
[44]
.
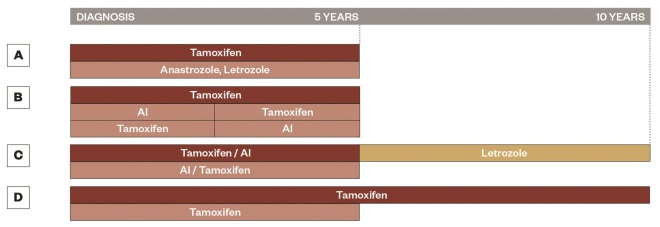
Figure 2: The evolution of adjuvant trial design
Schematic representation for the evolution of adjuvant trial design, A. represents the ATAC[34]
and BIG 1-98[40]
trials with anastrozole or letrozole alone for five years, B. represents the BIG 1-98[40]
and Intergroup Exemestane Study[41]
with two to three years of tamoxifen followed by an AI for two to three years or the reverse, C. represents the NCIC CTG MA.17 trial and NSABP B-42[42]
with five years of adjuvant tamoxifen (NSABP B-42 also allows five years of AI) followed by five years of letrozole, and D. represents the ATLAS[18]
and aTTom[44]
trials with the comparison of five versus ten years of tamoxifen.
In general, five years of adjuvant tamoxifen treatment, or an AI, achieved several clear results: longer (ten years) adjuvant anti-hormone therapy is better than shorter (five years) therapy for both disease-free survival and subsequent decreases in mortality; an AI alone has fewer life-threatening side effects than those associated with tamoxifen (i.e. endometrial cancer and thromboembolic effects)[45],
[46]
, but an AI alone causes an increased risk of osteoporotic fractures[46]
; AIs cause a 70% decrease in contralateral breast cancer compared with controls, whereas tamoxifen only causes a 50% decrease in contralateral breast cancer. The conclusion that an AI is superior to tamoxifen has recently been confirmed by an overview analysis of clinical trials by the Early Breast Cancer Trialists’ Collaborative Group in Oxford[46]
.
Long-term adjuvant anti-oestrogen therapy for premenopausal patients
One third of women who develop breast cancer do so during their premenopausal years[1]
. Premenopausal women are actively screened with mammography after the age of 40 years and optimal endocrine therapy is required for the patient with ER-positive disease. Tamoxifen has been the long-term adjuvant treatment of choice for premenopausal patients with ER-positive breast cancer for the past 25 years. Although tamoxifen causes a rise in circulating oestrogen levels[47]
, the fact that tamoxifen and its metabolites block the tumour ER is seen as a safety factor as it continues to halt oestrogen-stimulated tumour growth (see figure 1).
Combination cytotoxic chemotherapy is now routinely used as an adjuvant therapy for premenopausal breast cancer patients, but each different regimen used produces different rates of permanent amenorrhoea, which can be considered as an anti-hormonal ‘chemical oophorectomy’[48]
. As a rough guide, there are lower rates of amenorrhoea in patients aged under 30 years and higher rates in those aged over 40 years[49]
. It has been noted, for more than a decade, that very young women (<30 years) who develop breast cancer have a statistically higher rate of relapse despite receiving the same combination chemotherapy as older premenopausal women[50]
. Goldhirsch et al. recommended that “better treatments for very young women are required and may involve ovarian function suppression in addition to endocrine agents (tamoxifen or an AI) in patients with endocrine responsive tumours (i.e. ER-positive)”[50]
.
The clinical trials community has sought to improve endocrine therapy for premenopausal patients. Despite the fact that AIs are superior to tamoxifen in postmenopausal patients, AIs alone cannot be used to treat patients during their premenopausal years. This is because an initial drop of ovarian oestrogen production caused by the AI is compensated for by an increased secretion of gonadotropins from the pituitary gland. As a result, an AI alone cannot decrease circulating oestrogen levels effectively. Current clinical trials using ovarian function suppression (OFS) with a gonadotropin-releasing hormone superagonist (GnRH) to prevent gonadotropin release have addressed the question of whether reducing circulating oestrogen will improve the patient’s response to tamoxifen treatment alone or whether the approach of OFS can be used safely to devise a strategy to use an AI (see figure 1). The Suppression Of Ovarian Function Trial (SOFT)[51]
and the Tamoxifen and EXemestane Trial (TEXT)[52]
compared tamoxifen alone versus OFS plus tamoxifen versus OFS plus exemestane in patients with ER-positive tumours who remained premenopausal following adjuvant chemotherapy. Only those patients with continuing cycles needed OFS. The investigators concluded that the AI plus OFS significantly reduced reoccurrence at 68 months compared with tamoxifen plus OFS[51],
[52]
. However, as noted by Colleoni and Munzone, adverse events such as osteoporotic fractures, vaginal dryness, musculoskeletal symptoms, dyspareunia, and decreased libido, occurred more frequently with the AI[53]
.
Improvements in mortality rates after long-term anti-hormone therapy is stopped
The MA-17 trial is a large, randomised, double-blind, placebo-controlled study that was designed to evaluate the efficacy and safety profile of five further years of letrozole, or no treatment, in postmenopausal patients who were disease-free after five years of tamoxifen. The results of MA-17 demonstrated that letrozole is superior to placebo in improving disease-free survival and also showed that there was a profound reduction of 42% in the risk of reoccurrence regardless of the patient’s nodal status or prior chemotherapy exposure. There was also a significant decrease of 40% in the risk of distant metastasis, and an improvement in overall survival in women with node-positive disease. A reduction in mortality of 39% was noted among the approximately 2,500 women in the trial with node-positive disease[42],
[43]
. This was valid early evidence for extending long-term adjuvant anti-hormone therapy to aid patients’ survival. However, it also raised the question of what is considered an appropriate treatment length for extended adjuvant therapy, or whether the treatment should be lifelong.
The extended follow-up of adjuvant clinical trials has been particularly important in order to quantify the long-term societal benefits of adjuvant anti-hormone therapy. A consistent clinical observation occurs with five years or more of adjuvant anti-hormone treatment (i.e. after tamoxifen is stopped): there is no recurrence of tumour growth[17],
[18],
[54]
, something that would be expected to occur once oestrogen was free to bind to the ER in remaining micrometastases. This was an unanticipated clinical result. Tamoxifen was originally classified as a competitive inhibitor of oestrogen action at the tumour ER[16]
: it causes a G1 blockade in the breast cancer cell cycle[55],
[56]
and does not exhibit cytotoxic actions. As a result of this hypothesis, it was reasoned that tamoxifen must remain present, perhaps indefinitely, to block the regrowth of oestrogen-stimulated micrometastases. However, the clinical data proved that the micrometastases are not stimulated to grow after five years of adjuvant tamoxifen therapy is stopped. Indeed, it was noted that decreases in mortality were improved after tamoxifen therapy was stopped at five years[17]
. This clinical observation not only occurs with tamoxifen, but also occurs with long-term adjuvant AI therapy[36],
[37]
. Based on laboratory research[22],
[57],
[58]
, it was proposed that long-term oestrogen deprivation causes the selection of new micro-metastatic breast cancer cell populations with acquired resistance to anti-hormone therapy[59]
. These cell populations, however, are vulnerable to physiologic oestrogen that does not stimulate growth but triggers apoptosis and tumour regression[59]
. This is the unanticipated cytotoxic component of long-term anti-hormone therapy[22],
[60]
that prevents tumour regrowth with oestrogen. This cytotoxic action of oestrogen-induced apoptosis[57],
[60]
is accepted as the mechanism behind the successful treatment of MBC with high-dose synthetic oestrogens in patients more than five years postmenopause[7]
. This phenomenon is most dramatically illustrated in the Adjuvant Tamoxifen Longer Against Shorter (ATLAS) trial[18]
that compared stopping at five years of tamoxifen, or continuing for another five years for a ten-year total of adjuvant tamoxifen therapy (see figure 2). The mortality decreases observed with tamoxifen are noted most dramatically during the ten years after a decade of tamoxifen treatment had stopped. A comparison during tamoxifen treatment and after tamoxifen treatment shows that during years five to nine of treatment, the relative risk (RR) of mortality is 0.97 (0.78–1.18); i.e. little benefit[38]
. However, in the decade after tamoxifen treatment had stopped, RR=0.71 (0.58–0.88), P =0.002); i.e. major survival gains. The Adjuvant Tamoxifen—To Offer More? (aTTom) trial had a similar design to ATLAS and during years five to nine of treatment, the RR=1.08 (0.85–Â1.38); i.e. little survival gains. However, in the decade after tamoxifen is stopped, RR=0.75 (0.63–0.9), P =0.007; i.e. major survival gains. A combined analysis of aTTom and ATLAS[38]
demonstrated no benefit of continuing tamoxifen in the five years after extending tamoxifen treatment to ten years, as the carry over effect for the first five years of tamoxifen treatment is strong; however, mortality after ten years of tamoxifen is RR=0.75 (0.65–0.86), P =0.00004.
The proposal that a woman’s own oestrogen is causing apoptosis following adjuvant therapy[22]
is currently being tested in the clinical trial referred to as Study Of Letrozole Extension (SOLE)[61]
. In the SOLE study, there are two arms of treatment following the recruitment of breast cancer patients who have already completed five years of adjuvant anti-hormone therapy. In one arm, patients receive an additional five years of adjuvant AI therapy (as in the NCIC CTG MA.17 trial), but this continuous therapy is being compared to an arm that only has nine months of letrozole treatment per year, with a three-month drug holiday that is repeated for four years[61]
. The final year is a full year of letrozole treatment. Results are anticipated during the next few years. These data may refine and enhance the early effectiveness of adjuvant therapeutics. Another potential benefit will be financial savings within the healthcare system if intermittent AI treatment proved to be superior to continuous AI therapy. The savings could be more than a year of therapy costs through the second five years of extended intermittent adjuvant treatment.
The following sections focus on current innovations in combination anti-hormone therapies being evaluated in trials of MBC to restrain acquired anti-hormone resistance, thereby controlling tumour regrowth. These data are the conceptual basis for future adjuvant clinical trials.
Combining new agents to contend with acquired resistance of anti-hormonal therapy
New agents that target different survival pathways, as well as ERs, may prevent the development of premature acquired resistance to anti-hormone therapy in MBC. Cyclin-dependent kinases (CDK) 4 and 6 inhibitors and mTOR inhibitors have been tested with anti-hormones to treat ER-positive breast cancer (see ‘Figure 3: Breast cancer cell growth and the action sites of drugs’). PALbociclib Ongoing trials in the Management of breast cAncer (PALOMA-1/TRIO-18, also known as Study 1003) phase II clinical trials[62]
evaluated the safety and efficacy of palbociclib in combination with letrozole for patients with advanced ER-positive, HER2-negative breast cancer as a first-line treatment. Palbociclib is an inhibitor of CDKs 4 and 6 that normally promotes transition of the cell cycle from the G1 phase to the S phase (see figure 3). The study concluded that using palbociclib in conjunction with letrozole significantly improved progression-free survival from 10.2 months for letrozole alone, to 20.2 months for the combination of letrozole plus palbociclib[62]
. Another phase III study evaluated the efficacy of palbociclib plus fulvestrant versus fulvestrant alone in patients with advanced hormone-receptor-positive, human epidermal growth factor receptor 2-negative breast cancer, who relapsed or whose cancer progressed on a prior endocrine therapy, but only used the suboptimal loading dose treatment regimen (see ‘Fulvestrant and the prospect of new orally-active SERDs’). Premenopausal or perimenopausal patients also received goserelin. The study demonstrated that palbociclib combined with fulvestrant resulted in longer progression-free survival periods than fulvestrant alone, with 9.2 months for the combination and 3.8 months for the fulvestrant alone[63]
. The incidence of side effects was over 50% grade three and four neutropaenia[62],
[63]
. Healthcare costs for palbociclib are more than US$10,000 per month.
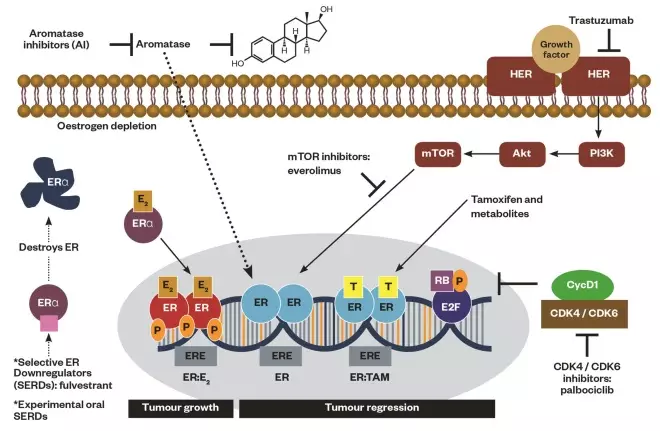
Figure 3: Breast cancer cell growth and the action sites of drugs
Schematic representation of breast cancer cell growth through oestrogen (i.e. oestradiol) binding to unoccupied oestrogen receptor (ERα) as a complex to the nucleus (tumour progression) and the action sites of drugs inhibiting or downregulating cellular growth at the receptor, cytoplasmic or nuclear level (tumour regression): mTOR inhibitors (i.e. everolimus) downregulating PI3K/AKT pathway, selective ER modulators (SERMs, tamoxifen and metabolites) blocking ER, selective ER downregulators (SERDs, fulvestrant) and experimental oral SERDs destroying ER, aromatase inhibitors (i.e. anastrozole, letrozole, exemestane) inhibiting aromatase enzyme, monoclonal antibody (i.e. trastuzumab) interfering with the HER2/neu receptor, cyclin-dependent kinase (CDK) 4/6 inhibitors (i.e. palbociclib) inhibiting CDK 4/6.
First-generation mTOR inhibitors (e.g. everolimus) target mTOR, which controls cell growth, proliferation, metabolism, and angiogenesis (see figure 3). The Breast Cancer Trials of OraL EveROlimus-2 (BOLERO-2) clinical trial[64]
combined everolimus and exemestane for women with advanced ER-positive/HER2-negative breast cancer who previously failed nonsteroidal AI therapy. Adding everolimus to exemestane in MBC in postmenopausal patients with ER-positive breast tumours achieved its goal of extending progression-free survival[65]
from 4.1 months in the exemestane arm to 10.6 months in the exemestane plus everolimus arm. However, everolimus had a greater number of grade three and four adverse events[66]
compared with exemestane alone. There was no overall improvement in the secondary end point of overall survival[67]
. However, the improvement in the median of progression-free survival led to its approval in the United States and Europe. It must be restated that BOLERO-2 recruited patients who had already failed nonsteroidal AI, and so patients were getting to the end of their anti-hormonal cascade. Everolimus costs more than US$11,000 per month.
Fulvestrant and the prospect of new orally-active SERDs
AIs and fulvestrant, with the latter a so-called ‘pure anti-oestrogen’, are effective agents for the treatment of acquired resistance to tamoxifen in MBC. This treatment paradigm followed translational laboratory research with a human breast cancer model of acquired resistance to tamoxifen in vivo to confirm the potential of using no oestrogen, or a pure anti-oestrogen, as a second-line therapy following the failure of tamoxifen treatment[20],
[21],
[68]
. A decade later, these laboratory studies were followed by two randomised clinical trials[69],
[70]
. Fulvestrant with a steroidal structure (see ‘Figure 4: SERDs’) binds with high affinity to the ER to induce an aberrant conformational change[71]
. The profound distortion of the ER complex makes the misshaped protein a target for rapid destruction by the cancer cell (see figure 4). The loss of the key signal transduction pathway for cell survival prevents tumour growth without any oestrogen-like effects. Fulvestrant’s pure antagonism, therefore, spares patients from the potentially dangerous oestrogen-like side effects of tamoxifen, especially the increases in endometrial cancer and thromboembolic events[72]
. Unfortunately, the original monthly 250mg intramuscular dosing used clinically has been a concern since fulvestrant’s initial approval by the Food and Drug Administration in 2002. This concern was addressed through the clinical trials mechanism. The Faslodex INvestigation of Dose evaluation in Estrogen Receptor-positive advanced breast cancer (FINDER) phase II trial[73]
randomised Western women, while the FINDER phase I trial[74]
randomised Japanese women in a parallel fashion. Both trials evaluated several end points for treating postmenopausal women with ER-positive locally–advanced, or reoccurring or progressing, MBC, after the failure of a prior endocrine therapy. Three fulvestrant dosing regimens were used: the approved dose (AD) of 250mg, 250mg and a loading dose (LD) during the first month, and a high dose (HD) of 500mg. The primary end point was objective response rate and the secondary end points were time to progression (TTP), rate of clinical benefit, tolerability, and pharmacokinetics. FINDER phase I and phase II clinical trials concluded that HD results in improved efficacy, a significant reduction in ER expression, and doubling the objective response rate. Also, the median TTP was 6.0 months, 7.5 months and 6.0 months for AD, LD and HD, respectively. The phase III COmparisoN of Faslodex In Recurrent or Metastatic Breast Cancer (CONFIRM) clinical trial[75],
[76]
randomised the same category of women as in the FINDER trial but pitted the HD fulvestrant (500mg regimen) against the AD (fulvestrant 250mg per month). Final results of the CONFIRM trial demonstrated a 19% reduction in risk of death, a 4.1-month difference in median overall survival, no increase in toxicity, and a statistically-significant increase in progression-free survival with the HD of 500mg when compared with 250mg (hazard ratio=0.80; 95% CI 0.68–0.94; P =0.006), corresponding to a 20% reduction in risk of progression.
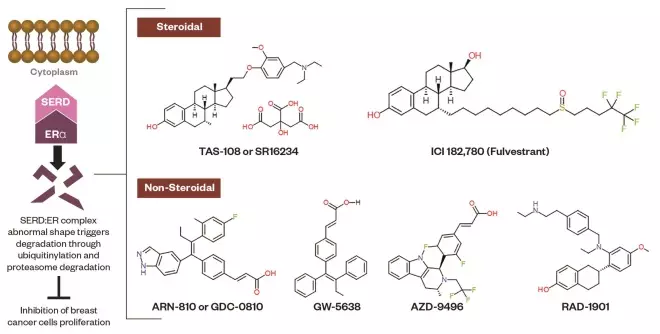
Figure 4: Selective estrogen receptor downregulators (SERDs)
Schematic representation of SERDs. The molecular mechanism of SERDs lies in binding to the ER, then the SERD:ER complex aberrant conformational change destroys the ER through the ubiquitin-proteasome system. The chemical structures of steroidal and non-steroidal SERDs are shown.
The next step forward was to study the impact of combining fulvestrant with AIs in clinical trials. The scientific concept was to destroy elevated ER levels caused by AI treatment and then to determine patient benefit. Results from different trials – Fulvestrant and Anastrozole Combination Therapy (FACT)[77]
, Study Of Faslodex vs Exemestane with/without Arimidex (SoFEA)[78]
, and the SouthWestern Oncology Group (SWOG s0226) – were confusing[79]
. However, all trials used only the LD regimen of fulvestrant followed by the AD of 250mg monthly (i.e. not HD fulvestrant). The FACT trial, as a first-line therapy for patients with receptor-positive postmenopausal breast cancer[77]
, compared anastrozole alone with a combination of fulvestrant and anastrozole. The FACT trial showed no clinical advantage for fulvestrant plus anastrozole over anastrozole alone. The SoFEA trial, as a second-line therapy for postmenopausal women with advanced hormone-receptor-positive breast cancer[78]
, randomised the same category of women in FACT with locally-advanced or metastatic disease, who relapsed or progressed on nonsteroidal AI. Women were distributed into three arms: fulvestrant plus anastrozole; fulvestrant plus placebo; and exemestane alone. The goal was to assess the use of fulvestrant in combination with continuous oestrogen deprivation. The SoFEA trial demonstrated no clinical advantage of 250mg fulvestrant with oestrogen deprivation over fulvestrant alone or exemestane alone after the loss of response to nonsteroidal AIs. It was concluded, in an accompanying editorial, that a combination regimen of fulvestrant and AIs could be best for women with no prior exposure to anti-oestrogen therapy in the adjuvant setting[80]
.
In contrast, the SWOG s0226 trial as first-line therapy for postmenopausal women with breast cancer,[79]
using the same LD approach maintained with the AD for fulvestrant, found that the combination of fulvestrant with the AI (anastrozole) was superior to anastrozole alone. Overall survival for the combination was 47.7 months compared with 41.3 months for anastrozole alone (P =0.05). Overall, clinical recommendations could be hard to define, but clearly the SWOG 0226 trial had a population of patients with better prognosis.
The Evaluation of Faslodex vs Exemestane Clinical Trial (EFECT) study[81]
evaluated the LD in postmenopausal women with advanced ER-positive breast cancer who had experienced progression or reoccurrence during treatment with a nonsteroidal AI. It was concluded that fulvestrant and exemestane LD is efficacious and well-tolerated. SoFEA supplements the results of EFECT; however, SoFEA[80]
differs from FACT[77]
and SWOG s0226[79]
by the population studied and the comparators. In the FACT study[77]
, the TTP in the fulvestrant plus anastrozole and anastrozole alone arms was no different. However, in the SWOG s0226 study[79]
, significant differences between the regimens, regarding progression-free survival and overall survival, were reported. The safety and efficacy of HD fulvestrant has been evaluated in the Fulvestrant fIRst-line Study Trial (FIRST), which concluded that fulvestrant HD dramatically prolongs TTP compared with anastrozole[82]
. Finally, in the neoadjuvant setting, data from the Neoadjuvant Endocrine Therapy for Women with Estrogen-Sensitive Tumors (NEWEST) trial[83]
demonstrated that fulvestrant HD reduced ERs and the proliferation biomarker Ki67 to a dramatically greater extent than the AD arm. The results from NEWEST support the findings of FINDER2, where AD, LD, and HD had similar efficacy but TTP was shorter with fulvestrant AD (3.1 months) compared with LD and HD arms (6.1 and 6.0 months, respectively)[84]
.
Medicinal chemists aim to optimise the pharmacological impact of fulvestrant to resolve concerns regarding its intramuscular route of administration, its poor solubility, and inadequate pharmacokinetics, which lead to incomplete receptor blockade. The development of an orally-active ‘pure anti-oestrogen’ appears to be a reasonable strategy[85]
. The new group of medicines under investigation is now called SERDs[86]
(see figure 4). However, here lies a pharmacological confusion that deserves discussion and consideration. The issue focuses on the true nomenclature between SERDs and the established pharmacology of medicines called Selective ER Modulators (SERMs), used ubiquitously in women’s health[87]
. By definition, fulvestrant is the pioneering SERD that has no oestrogen-like activity, at a heavily oestrogen-targeted site, because the shape of the ligand destroys the ER complex completely (see see figure 4). Compound GW5638 (see figure 4) is a newly discovered pioneering triphenylethylene[88]
with a novel acrylic acid side chain instead of the alkylaminoethoxy side chain that is the hallmark of SERMs[87]
. GW5638 is metabolically 4-hydroxylated to GW7604 in the same way that tamoxifen is metabolically activated to 4-hydroxytamoxifen. However, unlike tamoxifen, GW7608 triggers the destruction of the tumour cell ER complex[89],
[90],
[91]
. Destruction of the ER is clearly an advantage as it prevents the promiscuous oestrogen-like behaviour of the tamoxifen:ER complex. The AIs cause oestrogen deprivation that results in up-regulation and accumulation of the unoccupied ER. This biological consequence of AI action creates an opportunity for acquired resistance to AIs with the selection of cells with novel ER complexes that are activated without a ligand[25]
. As a result of mutations in the ER, which occur by trial and error, the cancer cell survives and MBC grows despite AI therapy. However, in the specific case of GW5638, which destroys the ER protein, the molecule retains full SERM activity elsewhere in the body (i.e. oestrogen-like effects on bones and lipids[86]
with only weak uterine stimulation). In the specific case of GW5638, it may be a strategic advantage to create a new therapy that is actually a ‘pseudo-SERD’, which destroys the ER but retains SERM action in other oestrogen target sites. This then means that since the acrylic side chain appears in the structures of other potential SERDs (see figure 4), the pharmacology and classification of these compounds must be vigorously evaluated. The primary goal, after all, is to develop a true orally-active ‘pure anti-oestrogen’ designed to replicate the pharmacology of the pioneering SERD, fulvestrant[27]
.
The putative SERDs, illustrated in figure 4, are still undergoing clinical development. ARN-810 is an orally-active SERD that demonstrates profound anti-tumour activity when tested in tamoxifen-resistant cancer xenografts[92]
. AZD-9496, a potent orally-bioavailable SERD, was highly effective in MCF-7 breast cancer cells in an endocrine sensitive model and in long-term oestrogen-deprived breast cancer cells as a resistant model[93],
[94]
. RAD-1901 is another agent that showed safety and efficacy for the treatment of vasomotor symptoms in healthy postmenopausal women[95],
[96]
. Finally, TAS-108, an orally-active steroid, has demonstrated good patient tolerance and clinical benefit in phase I and phase II trials in sequence[97],
[98]
.
Future advances to improve adjuvant anti-hormone therapy
During the next 15 years, it is estimated that there will be a 50% increase in the incidence of breast cancer. Specifically, this will be ER-positive breast cancer caused by the population having an increased lifespan[99]
; it is already known that the incidence of ER-positive breast cancer correlates with an increase in age. At present, the distribution of the ER in breast cancer is about 80% ER-positive and 20% ER-negative, but it is estimated that the distribution will become 95% ER-positive with only 5% ER-negative[99]
. Strategies to prevent or cure ER-positive breast cancer are an important priority in healthcare.
The fact that long-term adjuvant anti-hormone therapy has been so successful in maintaining a disease-free state for up to 50% ER-positive breast cancer patients for decades is an important first step for the future[18]
. Breast cancer has been converted from the fatal disease of just 40 years ago to being either cured or transformed into a chronic disease with a good prognosis. Tamoxifen, the pioneering targeted therapeutic agent for long-term anti-oestrogenic therapy, controlled the disease and saved lives in a cost-effective manner. The refinement of AI adjuvant therapy for postmenopausal patients has further improved breast cancer maintenance with fewer side effects and increased efficacy[45],
[46]
. The goal for the next few decades is to improve survival, rationally, by subverting acquired tumour anti-hormone resistance.
Current molecular medicine has made a valuable start by improving the prognosis of anti-hormone resistant ER-positive MBC, using either an inhibitor of mTOR or a CD4/6 inhibitor. Treatment advances in MBC can be predicted to enhance survival in the adjuvant setting because the low tumour burden of micrometastases is already a good predictor of therapeutic success. However, the challenge with the current adjuvant maintenance strategy that must be continued for a decade is with patient compliance to a new multi-drug adjuvant cocktail that must be taken for that long. Compliance with a cheap medicine, such as tamoxifen mono-adjuvant therapy, is already a concern; compliance decreases to 60% over five years and the lives lost can be calculated[100]
. The future prospect of anti-hormone therapies that have relatively few life-threatening side effects (such as AIs), or an inconvenient route of administration (such as fulvestrant), being supplemented with either an mTOR inhibitor or a CD4/6 inhibitor in a new long-term adjuvant therapy cocktail represents a challenge because both the mTOR inhibitor everolimus[66]
and CDK4/6 inhibitor pablociclib[62],
[63]
profoundly increased serious side effects – grade three and four toxic adverse events – in adjuvant trials. Palbociclib increased grade three and four neutropaenia in half of the patients taking palbociclib plus letrozole[62],
[63]
. Carey and Perou[101]
noted that not only are there increases in side effects, but also two promising biomarkers (the loss of p16ink4a and gain in cyclin D1) as possible markers to predict efficacy with palbociclib in ER-positive breast cancer, failed to predict any benefit[70]
. Here there has been a failure of precision medicine. The rationale of adding palbociclib to all ER-positive breast cancer patients on any AI/CDK4/6 adjuvant trial for a decade would not only be self-defeating with rapid decreases in compliance within a year or two, but also would increase healthcare costs enormously as the medicine costs US$10,000 per month. Obviously, another way must be considered to advance the clear-cut gains in survival that using long-term anti-oestrogen therapy brings, until the precision medicine for the individual can be seen to work effectively in saving lives.
Acquired resistance to long-term adjuvant endocrine therapy with AIs[102]
occurs because of growth factor-mediated mechanisms or mutations that occur in the ER. Mutations in the ER permit constitutive activation of the unoccupied ER and tumour growth[25]
. The use of orally-active ‘pure anti-oestrogens’ to replace AIs could be the appropriate solution to the mutation resistance mechanism because it destroys the ER. With no ER, there would not be any mutations or uncontrolled growth. However, this strategy may also increase the probability of a shift towards ER-negative breast cancer. The tumours would become ER-negative, hormone independent, and rapidly fatal. The current experience with fulvestrant in clinical studies with MBC suggests that this hypothesis is not true in patients, so a trial of fulvestrant in the adjuvant setting may be appropriate. Compliance would be a problem with a HD fulvestrant regimen, but this would swiftly encourage the development of either an orally-active ‘true SERD’ or an orally-active ‘pseudo SERD’ as important additions to the therapeutic armamentarium, to replace both tamoxifen and AIs.
A true orally-active SERD may provide better control of breast cancer reoccurrence than the AI adjuvant strategy. The hypothesis to be tested would be that the destruction of the pool of the unoccupied ERs, expanded and enhanced by ER gene amplification during AI adjuvant therapy, would be destroyed by the SERD. The mechanism for a significant population of AI-acquired resistance, through ER mutations, would be controlled. By contrast, if the new orally-active compound was a ‘pseudo-SERD’, like GW5638, that could reduce ER mutations in micrometastatic breast cancer, then the same process would occur as a true SERD, and ER mutations would not occur to cause acquired resistance. The advantage of a ‘pseudo-SERD’ is that its pharmacology is not pure anti-oestrogen everywhere; rather, it acts like a SERM. The ‘pseudo-SERD’ would also enhance women’s health by reducing coronary heart disease, reducing osteoporotic fractures, and improving gynaecological health[87]
. All the quality of life issues that occur with AIs could be solved by an agent used as a ten-year adjuvant therapy. Compliance could be maintained with the knowledge that, not only was the ‘pseudo-SERD’ improving the patient’s prospects of surviving breast cancer, but it was also enhancing their general health by preventing osteoporosis, coronary heart disease, and improving their general quality of life.
Women should not have the only option of “there is no anti-oestrogen, like no oestrogen at all”, as we have with an AI, but should have the advantage of precision medicine at different target sites around their body.
Balkees Abderrahman is postdoctoral fellow, breast medical oncology and V. Craig Jordan is Dallas/Ft. Worth living legend chair of cancer research, professor of breast medical oncology, professor of molecular and cellular opncology, chief of the section of basic science research and pharmacology, University of Texas MD Anderson Cancer Center, 1500 Holcombe Blvd. Unit #1354, Houston, TX 77030, United States. Correspondence to:
vcjordan@mdanderson.org
Acknowledgments
This article is dedicated to three individuals whose contributions continue to enhance the treatment of breast cancer and improve women’s health: Angela Brodie, Harry Brodie and Aron Goldhirsch.
Author disclosure and conflicts of interest
The work reported here was supported by the National Institutes of Health (NIH), MD Anderson’s Cancer Center support grant CA016672 and Susan G Komen for the Cure Foundation under award number SAC100009, and Cancer Prevention Research Institute of Texas (CPRIT) for the STARs and STARs Plus Awards. V Craig Jordan thanks the benefactors of the Dallas/Ft Worth Living Legend Chair of Cancer Research for their generous support. No writing assistance was utilised in the production of this manuscript.
Reading this article counts towards your CPD
You can use the following forms to record your learning and action points from this article from Pharmaceutical Journal Publications.
Your CPD module results are stored against your account here at The Pharmaceutical Journal. You must be registered and logged into the site to do this. To review your module results, go to the ‘My Account’ tab and then ‘My CPD’.
Any training, learning or development activities that you undertake for CPD can also be recorded as evidence as part of your RPS Faculty practice-based portfolio when preparing for Faculty membership. To start your RPS Faculty journey today, access the portfolio and tools at www.rpharms.com/Faculty
If your learning was planned in advance, please click:
If your learning was spontaneous, please click:
References
[1] Cancer Research UK. Cancer statistics for the UK. Available at: http://www.cancerresearchuk.org/health-professional/cancer-statistics (accessed March 2016).
[2] Peto R, Boreham J, Clarke M et al. UK and USA breast cancer deaths down 25% in year 2000 at ages 20–69 years. The Lancet 2000;355:1822. doi: 10.1016/S0140-6736(00)02277-7
[3] Berry DA, Inoue L, Shen Y et al. Modeling the impact of treatment and screening on US breast cancer mortality: a Bayesian approach. J Natl Cancer Inst Monogr 2006:30–36. doi: 10.1093/jncimonographs/lgj006
[4] Narod SA, Iqbal J & Miller AB. Why have breast cancer mortality rates declined? J Cancer Policy 2015;5:8–17. doi: 10.1016/j.jcpo.2015.03.002
[5] Jordan VC. A century of deciphering the control mechanisms of sex steroid action in breast and prostate cancer: the origins of targeted therapy and chemoprevention. Cancer Res. 2009;69:1243–1254. doi: 10.1158/0008-5472.CAN-09-0029
[6] Jordan VC. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer 2014;21:R235–R246. doi: 10.1530/ERC-14-0092
[7] Haddow A, Watkinson JM, Paterson E et al. Influence of synthetic oestrogens on advanced malignant disease. Br Med J 1944;2:393–398. PMCID: PMC2286289
[8] Glascock RF & Hoekstra WG. Selective accumulation of tritium-labelled hexoestrol by the reproductive organs of immature female goats and sheep. Biochem J 1959;72:673–682. PMCID: PMC1196992
[9] Jensen EV & Jacobson HI. Basic guides to the mechanism of estrogen action. Recent Prog Horm Res. 1962;XVIII:387–414. Pincus, G., editor., ed. New York: and London: Academic Press.
[10] Folca PJ, Glascock RF & Irvine WT. Studies with tritium-labelled hexoestrol in advanced breast cancer. Comparison of tissue accumulation of hexoestrol with response to bilateral adrenalectomy and oophorectomy. The Lancet 1961;2:796–798. doi: 10.1016/S0140-6736(61)91088-1
[11] Jensen E, Block G, Smith S et al. Estrogen receptors and breast cancer response to adrenalectomy. J Natl Cancer Inst Monogr 1971;34 55–70. PMID: 5140877
[12] McGuire WL, Carbone PP & Vollmer EP (ed.). Estrogen receptors in human breast cancer. New York: Raven Press. 1975;1–7.
[13] Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nature Reviews Drug Discovery. 2003;2:205–213. doi: 10.1038/nrd1031
[14] Jordan VC & Brodie AM. Development and evolution of targeted endocrine therapies for the treatment of breast cancer. Steroids 2007;72:7–25. doi: 10.1016/j.steroids.2006.10.009
[15] Sledge GW, Mamounas EP, Hortobagyi GN et al. Past, present, and future challenges in breast cancer treatment. J Clin Oncol 2014;32:1979–1986. doi: 10.1200/JCO.2014.55.4139
[16] Jordan VC. Biochemical pharmacology of antiestrogen action. Pharmacological Reviews 1984;36(4):245–276. PMID: 6395141
[17] Early Breast Cancer Trialists’ Collaborative Group. Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. The Lancet 1998;351(9114):1451–1467. doi: 10.1016/S0140-6736(97)11423-4
[18] Davies C, Pan H, Godwin J et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. The Lancet 2013;381:805–816. doi: 10.1016/S0140-6736(12)61963-1
[19] Gottardis MM, Robinson SP & Jordan VC. Estradiol-stimulated growth of MCF-7 tumors implanted in athymic mice: a model to study the tumoristatic action of tamoxifen. J Steroid Biochem 1988;30(1–6):311–314. PMID: 3386259
[20] Gottardis MM & Jordan VC. Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration. Cancer Res 1988;48:5183–5187. PMID: 3409244
[21] Gottardis MM, Wagner RJ, Borden EC et al. Differential ability of antiestrogens to stimulate breast cancer cell (MCF-7) growth in vivo and in vitro. Cancer Res 1989;49:4765–4769.
[22] Yao K, Lee ES, Bentrem DJ et al. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res 2000;6:2028–2036. PMID: 10815929
[23] Fan P, Agboke FA, McDaniel RE et al. Inhibition of c-Src blocks estrogen-induced apoptosis and restores estrogen-stimulated growth in long-term estrogen-deprived breast cancer cells. Eur J Cancer 2014;50:457–468. doi: 10.1016/j.ejca.2013.10.001
[24] Fan P, Agboke FA, Cunliffe HE et al. A molecular model for the mechanism of acquired tamoxifen resistance in breast cancer. Eur J Cancer 2014;50:2866–2876. doi: 10.1016/j.ejca.2014.08.011
[25] Jordan VC, Curpan R & Maximov PY. Estrogen receptor mutations found in breast cancer metastases integrated with the molecular pharmacology of Selective ER Modulators. J Natl Cancer Inst 2015;107:djv075. doi: 10.1093/jnci/djv075
[26] Santen RJ, Brodie H, Simpson ER et al. History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev 2009;30:343–375. doi: 10.1210/er.2008-0016
[27] Jordan VC. The only true antiestrogen is no estrogen. Mol Cell Endocrinol 1990;74;C91–C95. doi: 10.1016/0303-7207(90)90220-3
[28] Brodie AMH, Schwarzel WC, Shaikh AA et al. The effect of an aromatase inhibitor, 4-hydroxy-androstene-3,17-dione, on estrogen-dependent processes in reproduction and breast cancer. Endocrinol 1977;100:1684–1695. doi: 10.1210/endo-100-6-1684
[29] Brodie AMH, Garrett WM, Hendrickson JR et al. Inactivation of aromatase activity in placental and ovarian microsomes by 4-hydroxyandrostene-3,17-dione and 4-acetoxyandrostene-3,17-dione. Steroids 1981;38:693–702.
[30] Coombes RC, Goss P, Dowsett M et al. 4-Hydroxyandrostenedione in treatment of postmenopausal patients with advanced breast cancer. The Lancet 1984;2:1237–1239. doi: 10.1016/S0140-6736(84)92795-8
[31] Goss PE, Powles TJ, Dowsett M et al. Treatment of advanced postmenopausal breast cancer with an aromatase inhibitor, 4-Hydroxyandrostenedione: phase II report. Cancer Res 1986;46:4823–4826.
[32] Consensus conference. Adjuvant chemotherapy for breast cancer. JAMA 1985;254:3461–3463. doi: 10.1001/jama.1985.03360240073038
[33] Fisher B, Dignam J, Bryant J et al. Five versus more than five years of tamoxifen therapy for breast cancer patients with negative lymph nodes and estrogen receptor-positive tumors. J Natl Cancer Inst 1996;88:1529–1542. doi: 10.1093/jnci/88.21.1529
[34] Baum M, Budzar AU, Cuzick J et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. The Lancet 2002;359:2131–2139. doi: 10.1016/S0140-6736(02)09088-8
[35] Howell A, Cuzick J, Baum M et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. The Lancet 2005;365:60–62. doi: 10.1016/S0140-6736(04)17666-6
[36] Arimidex, Tamoxifen, Alone or in Combination (ATAC) Trialists’ Group: Forbes JF, Cuzick J et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-month analysis of the ATAC trial. Lancet Oncol 2008;9:45–53. doi: 10.1016/S1470-2045(07)70385-6
[37] Cuzick J, Sestak I, Baum M et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol 2010;11:1135–1141. doi: 10.1016/S1470-2045(10)70257-6
[38] Schiavon G & Smith IE. Status of adjuvant endocrine therapy for breast cancer. Breast Cancer Res 2014;16:206. doi: 10.1186/bcr3636
[39] Blok EJ, Derks MGM, van der Hoeven JJM et al. Extended adjuvant endocrine therapy in hormone-receptor positive early breast cancer: current and future evidence. Cancer Treat Rev 2015;41:271–276. doi: 10.1016/j.ctrv.2015.02.004
[40] The Breast International Group (BIG) 1-98 Collaborative Group. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med 2005;353:2747–2757. doi: 10.1056/NEJMoa052258
[41] Coombes RC, Hall E, Gibson LJ et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med 2004;350:1081–1092. doi: 10.1056/NEJMoa040331
[42] Goss PE, Ingle JN, Martino S et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med 2003;349(19):1793–1802. doi: 10.1056/NEJMoa032312
[43] Goss PE, Ingle JN, Martino S et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Cancer Inst 2005;97:1262–1271. doi: 10.1093/jnci/dji250
[44] Gray RG, Rea D, Handley K et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013;18(5). Available at: http://meetinglibrary.asco.org/content/112995-132 (accessed May 2016).
[45] Dowsett M, Cuzick J, Ingle J et al. Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J Clin Oncol 2010;28:509–518. doi: 10.1200/JCO.2009.23.1274
[46] Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), writing committee: Dowsett M et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. The Lancet 2015;386:1341–1352. doi: 10.1016/S0140-6736(15)61074-1
[47] Jordan VC, Fritz NF, Langan-Fahey S et al. Alteration of endocrine parameters in premenopausal women with breast cancer during long-term adjuvant therapy with tamoxifen as the single agent. J Natl Cancer Inst 1991;83:1488–1491. doi: 10.1093/jnci/83.20.1488
[48] Jordan VC. Chemotherapy is anti-hormonal therapy – how much proof do oncologists need? Eur J Cancer 1998;34:606–608. doi: 10.1016/S0959-8049(98)00040-9
[49] Azim Jr HA, Azambuja ED, Colozza M et al. Long-term toxic effects of adjuvant chemotherapy in breast cancer. Ann Oncol 2011;22:1939–1947. doi: 10.1093/annonc/mdq683
[50] Goldhirsch A, Gelber RD, Yothers G et al. Adjuvant therapy for very young women with breast cancer: need for tailored treatments. J Natl Cancer Inst Monogr 2001;(30):44–51. PMID: 11773291
[51] Pagani O, Regan MM, Walley BA et al. Adjuvant exemestane with ovarian suppression in premenopausal breast cancer. N Engl J Med 2014;371:107–118. doi: 10.1056/NEJMoa1404037
[52] Francis PA, Regan MM, Fleming GF et al. Adjuvant ovarian suppression in premenopausal breast cancer. N Engl J Med 2015;372:436–446. doi: 10.1056/NEJMoa1412379
[53] Colleoni M & Munzone E. Picking the optimal endocrine adjuvant treatment for pre-menopausal women. The Breast 2015;2:S11–14. doi: 10.1016/j.breast.2015.07.004
[54] Early Breast Cancer Trialists’ Collaborative Group (EBCTCG): Davies C, Godwin J et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. The Lancet 2011;378:771–784. doi: 10.1016/S0140-6736(11)60993-8
[55] Osborne CK, Boldt DH, Clark GM et al. Effects of tamoxifen on human breast cancer cell cycle kinetics: accumulation of cells in early G1 phase. Cancer Res 1983;43:3583–3585. PMID: 6861130
[56] Sutherland RL, Hall RE & Taylor IW. Cell proliferation kinetics of MCF-7 human mammary carcinoma cells in MCF-7 in culture and effects of tamoxifen on exponentially growing and plateau-phase cells. Cancer Res 1983;43:3998–4006. PMID: 6871841
[57] Song RX, Mor G, Naftolin F et al. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17beta-estradiol. J Natl Cancer Inst 2001;93(22):1714–1723. doi: 10.1093/jnci/93.22.1714
[58] Lewis JS, Meeke K, Osipo C et al. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst 2005;97:1746–1759. doi: 10.1093/jnci/dji400
[59] Jordan VC. Linking estrogen-induced apoptosis with decreases in mortality following long-term adjuvant tamoxifen therapy. J Natl Cancer Inst 2014;106:dju296. doi: 10.1093/jnci/dju296
[60] Jordan VC. The 38th David A. Karnofsky lecture: the paradoxical actions of estrogen in breast cancer–survival or death? J Clin Oncol 2008;26:3073–3082. doi: 10.1200/JCO.2008.17.5190
[61] Jordan VC & Ford LG. Paradoxical clinical effect of estrogen on breast cancer risk: a “new” biology of estrogen-induced apoptosis. Cancer Pre Res 2011;4:633–637. doi: 10.1158/1940-6207.CAPR-11-0185
[62] Finn RS, Crown JP, Lang I et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3
[63] Turner NC, Ro J, André F et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med 2015;373:209–219. doi: 10.1056/NEJMoa1505270
[64] Baselga J, Campone M, Piccart M et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520–529. doi: 10.1056/NEJMoa1109653
[65] Yardley DA, Noguchi S, Pritchard KI et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther 2013;30:870–884. doi: 10.1007/s12325-013-0060-1
[66] Beaver JA & Park BH. The BOLERO-2 trial: the addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol 2012;8:651–657. doi: 10.2217/fon.12.49
[67] Piccart M, Hortobagyi GN, Campone M et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2†. Ann Oncol 2014;25:2357–2362. doi: 10.1093/annonc/mdu456
[68] Gottardis MM, Jiang SY, Jeng MH et al. Inhibition of tamoxifen-stimulated growth of an MCF-7 tumor variant in athymic mice by novel steroidal antiestrogens. Cancer Res 1989;49:4090–4093. PMID: 2743303
[69] Osborne CK, Pippen J, Jones SE et al. Double blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole inpostmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol 2002;20:3386–3395. doi: 10.1200/JCO.2002.10.058
[70] Howell A, Robertson JF, Quaresma AJ et al. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breastcancer progressing after prior endocrine treatment. J Clin Oncol 2002;20:3396–3403. doi: 10.1200/JCO.2002.10.057
[71] Wijayaratne AL & McDonnell DP. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem 2001;276:35684–35692. PMID: 11473106
[72] Jones SE. A new estrogen receptor antagonist—an overview of available data. Breast Cancer Res Treat 2002;75:S19–21; discussion S33–35. PMID: 12353819
[73] Pritchard KI, Rolski J, Papai Z et al. Results of a phase II study comparing three dosing regimens of fulvestrant in postmenopausal women with advanced breast cancer (FINDER2). Breast Cancer Res Treat 2010;123:453–461. doi: 10.1007/s10549-010-1022-9
[74] Ohno S, Rai Y, Iwata H et al. Three dose regimens of fulvestrant in postmenopausal Japanese women with advanced breast cancer: results from a double-blind, phase II comparative study (FINDER1). Ann Oncol 2010;21:2342–2347. doi: 10.1093/annonc/mdq249
[75] Di Leo A, Jerusalem G, Petruzelka L et al. Results of the CONFIRM phase III trial comparing fulvestrant 250mg with fulvestrant 500mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol 2010;28:4594–4600. doi: 10.1200/JCO.2010.28.8415
[76] Di Leo A, Jerusalem G, Petruzelka L et al. Final overall survival: fulvestrant 500 mg vs 250 mg in the randomized CONFIRM trial. J Natl Cancer Inst 2014;106:djt337. doi: 10.1093/jnci/djt337
[77] Bergh J, Jönsson PE, Lidbrink EK et al. FACT: an open-label randomized phase III study of fulvestrant and anastrozole in combination compared with anastrozole alone as first-line therapy for patients with receptor-positive postmenopausal breast cancer. J Clin Oncol 2012;30:1919–1925. doi: 10.1200/JCO.2011.38.1095
[78] Johnston SR, Kilburn LS, Ellis P et al. Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): a composite, multicentre, phase 3 randomised trial. Lancet Oncol 2013;14:989–998. doi: 10.1016/S1470-2045(13)70322-X
[79] Mehta RS, Barlow WE, Albain KS et al. Combination anastrozole and fulvestrant in metastatic breast cancer. N Engl J Med 2012;367:435–444. doi: 10.1056/NEJMoa1201622
[80] Di Leo A & Malorni L. Polyendocrine treatment in estrogen receptor-positive breast cancer: a “FACT” yet to be proven. J Clin Oncol 2012;30:1897–1900. doi: 10.1200/JCO.2012.41.7394
[81] Chia S, Gradishar W, Mauriac L et al. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol 2008;26:1664–1670. doi: 10.1200/JCO.2007.13.5822
[82] Robertson JF, Llombart-Cussac A, Rolski J et al. Activity of fulvestrant 500 mg versus anastrozole 1mg as first-line treatment for advanced breast cancer: results from the FIRST study. J Clin Oncol 2009;20;27:4530–4535. doi: 10.1200/JCO.2008.21.1136
[83] Kuter I, Hegg R, Singer CF et al. Fulvestrant 500mg vs 250mg: first results from NEWEST, a randomized, phase II neoadjuvant trial in postmenopausal women with locally advanced, estrogen receptor-positive breast cancer. Breast Cancer Res Treat 2007;106:S7.
[84] Pritchard K, Rolski J, Pápai Z et al. A phase II study (FINDER 2) comparing three dosing regimens of fulvestrant in postmenopausal women with estrogen receptor-positive advanced breast cancer. Cancer Res 2009;69:4069–4095. doi: 10.1158/0008-5472.SABCS-09-4095
[85] Karuturi M, Abderrahman B, Hortobagyi GN et al. Oral pure antiestrogens as a solution to acquired drug resistance to aromatase inhibitors. Breast Cancer Manag 2015;4:275–277. doi: 10.2217/bmt.15.22
[86] McDonnell DP, Wardell SE & Norris JD. Oral Selective Estrogen Receptor Downregulators (SERDs), a breakthrough endocrine therapy for breast cancer. J Med Chem 2015;58:4883–4887. doi: 10.1021/acs.jmedchem.5b00760
[87] Maximov PY, Lee TM & Jordan VC. The discovery and development of Selective Estrogen Receptor Modulators (SERMs) for clinical practice. Curr Clin Pharmacol 2013;8:135–155. doi: 10.2174/1574884711308020006
[88] Willson TM, Henke BR, Momtahen TM et al. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic acid: a non-steroidal estrogen with functional selectivity for bone over uterus in rats. J Med Chem 1994;37:1550–1552. doi: 10.1021/jm00037a002
[89] Willson TM, Norris JD, Wagner BL et al. Dissection of the molecular mechanism of action of GW5638, a novel estrogen receptor ligand, provides insights into the role of estrogen receptor in bone. Endocrinol 1997;138:3901–3911. doi: 10.1210/endo.138.9.5358
[90] Wijayaratne AL, Nagel SC, Paige LA et al. Comparative analysis of mechanistic differences among antiestrogens. Endocrinol 1999;140:5828–5840. doi: 10.1210/endo.140.12.7164
[91] Bentrem D, Dardes R, Liu H et al. Molecular mechanism of action at estrogen receptor alpha of a new clinically relevant antiestrogen (GW7604) related to tamoxifen. Endocrinol 2001;142:838–846. doi: 10.1210/endo.142.2.7932
[92] Lai A, Kahraman M & Govek S. Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts. J Med Chem 2015;58:4888–4904. doi: 10.1021/acs.jmedchem.5b00054
[93] De Savi C, Bradbury RH, Rabow AA et al. Abstract 3650: Discovery of the clinical candidate AZD9496: a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. Cancer Res 2015;75:3650–3650. doi: 10.1158/1538-7445.AM2015-3650
[94] De Savi C, Bradbury RH, Rabow AA et al. Optimization of a novel binding motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic Acid (AZD9496), a potent and orally bioavailable Selective Estrogen Receptor Downregulator and Antagonist. J Med Chem 2015;58:8128–8140. doi: 10.1021/acs.jmedchem.5b00984
[95] Garner F, Shomali M, Paquin D et al. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anti-cancer Drugs 2015;26:948–956. doi: 10.1097/CAD.0000000000000271
[96] Taylor HS. Designing the ideal selective estrogen receptor modulator–an achievable goal? Menopause 2009;16:609–615. doi: 10.1097/gme.0b013e3181906fa3
[97] Inaji H, Iwata H, Nakayama T et al. Randomized phase II study of three doses of oral TAS-108 in postmenopausal patients with metastatic breast cancer. Cancer Sci 2012;103:1708–1713. doi: 10.1111/j.1349-7006.2012.02354.x
[98] Buzdar A, Vogel C, Schwartzberg L et al. Randomized double-blind phase 2 trial of 3 doses of TAS-108 in patients with advanced or metastatic postmenopausal breast cancer. Cancer 2012;118:3244–3253. doi: 10.1002/cncr.26419
[99] Rosenberg PS, Barker KA, Anderson WF et al. Estrogen receptor status and the future burden of invasive and in-situ breast cancers in the United States. J Natl Cancer Inst 2015;107:djv159. doi: 10.1093/jnci/djv159
[100] McCowan C, Wang S, Thompson AM et al. The value of high adherence to tamoxifen in women with breast cancer: a community-based cohort study. Brit J Cancer 2013;109:1172–1180. doi: 10.1038/bjc.2013.464
[101] Carey LA & Perou CM. Palbociclib–Taking Breast-Cancer Cells Out of Gear. N Engl J Med 2015;373:273–274. doi: 10.1056/NEJMe1506680
[102] Fan P, Maximov PY, Curpan RF et al. The molecular, cellular and clinical consequences of targeting the estrogen receptor following estrogen deprivation therapy. Mol Cell Endocrinol 2015;418:245–263. doi: 10.1016/j.mce.2015.06.004
[103] International Breast Cancer Study Group (IBCSG): Castiglione-Gertsch M, O’Neill A et al. Adjuvant chemotherapy followed by goserelin versus either modality alone for premenopausal lymph node-negative breast cancer: a randomized trial. J Natl Cancer Inst 2003;95:1833–1846. doi: 10.1093/jnci/djg119
[104] Regan MM, Pagani O, Francis PA et al. Predictive value and clinical utility of centrally assessed ER, PgR, and Ki-67 to select adjuvant endocrine therapy for premenopausal women with hormone receptor-positive, HER2-negative early breast cancer: TEXT and SOFT trials. Breast Cancer Res Treat 2015;154:275–286. doi: 10.1007/s10549-015-3612-z