Key points:
- Side effects of statins, including muscle aches, diabetes and liver function test abnormalities, are increasingly recognised.
- Statin-related muscle side effects have recently been systematically classified.
- Rare autoimmune phenomena related to statin therapy, including necrotising myopathy, interstitial lung disease and lupus-like reactions, are now recognised.
- Statins increase the risk of transition to diabetes in susceptible individuals with the metabolic syndrome.
- There is little systematic evidence to link cognitive impairment with statin therapy.
Introduction
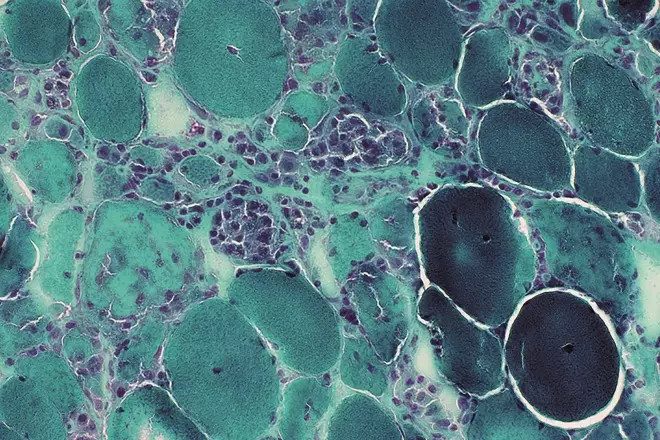
Image reproduced with kind permission of Professor Alan Pestronk, Neuromuscular Disease Center, Washington University, St. Louis, MO, USA
Statin therapy has demonstrated remarkable efficacy in reducing mortalities associated with cardiovascular diseases but reports on a variety of statin-associated side effects show myopathy as the most commonly reported side effect. In the image: a Gomori trichrome stain of very active, sub-acute myopathy associated with statins, showing muscle fibres in varying stages of necrosis, phagocytosis & regeneration
Statins, inhibitors of the 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase enzyme, are widely used in clinical practice as first-line treatment for dyslipidaemia and in the prevention of cardiovascular disease (CVD) events[1],
[2]
. Statins have been used for decades in the primary and secondary prevention of CVD among children and adults, males and females, diabetics and non-diabetics, who are at moderate to high risk of CVD[2]
. Statins are known to have a good safety record but are associated with side effects, the most common of which are related to muscle[3],
[4],
[5],
[6],
[7]
. The ubiquity of statin usage has focused attention on their side effects because, although relative rates of adverse events are low, the absolute numbers affected are high. The management of statin-related side effects comprises 10–25% of the workload of lipid clinics and, therefore, has been subject to the production of specific guidelines, such as Canadian[8],
[9]
and European guidelines[6]
for diagnosis and management of statin adverse events, and systematic reviews of the evidence base, such as those conducted by the US National Lipid Association (NLA)[10]
. This article provides an update on the current literature and international consensus regarding the safety profile of statins and their drug interactions, as well as the incidence, pathophysiology, diagnosis and management of muscular and non-muscular side effects of statins.
Sources and selection criteria
To identify scientific studies for this review, searches were performed on PubMed/MEDLINE using combinations of the search terms ‘statin’, ‘side effect’ and ‘muscle’, ‘liver function’, ‘pancreatitis’, ‘diabetes’, and ‘brain’ from 2000 to 2015 inclusive. There were no exclusion criteria. Other articles, technical reports and documents cited in the reference lists of publications were identified and located on organisation websites (such as those of the Cochrane Library, World Health Organization and Public Health England) via Google search.
Adverse effects of statins
Mechanistic effects of statins on mitochondria and programmed cell death (apoptosis)
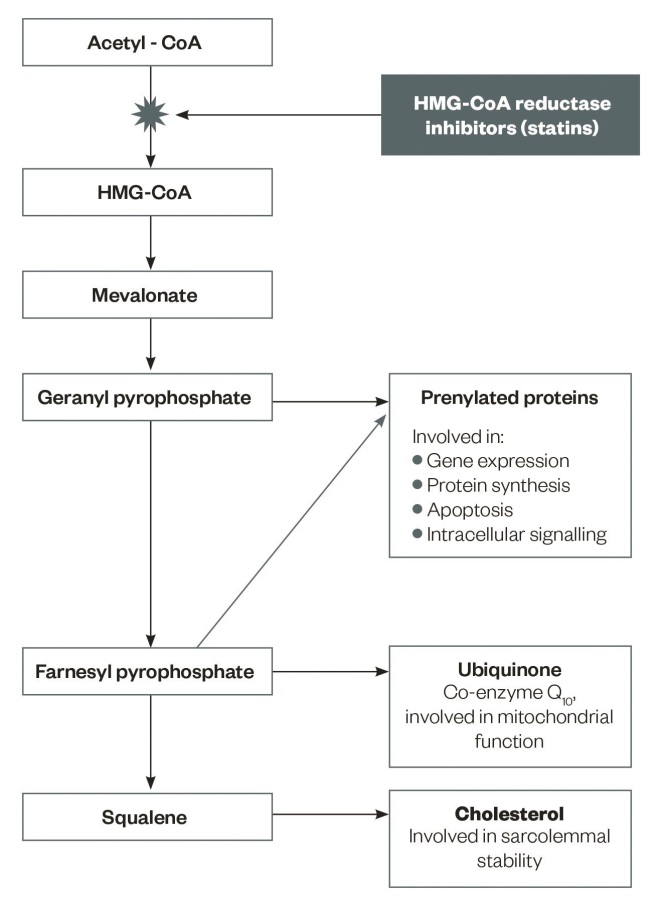
Figure 1: Metabolic pathways and products potentially affected by statins
Adapted from Stock J. Atherosclerosis 2015;242:346–350.
Intermediates in cholesterol synthesis and their role in cellular processes
Statins impair the metabolic pathway of cholesterol synthesis by blocking the production of mevalonate, a rate-limiting step in the biosynthesis of cholesterol[11]
. Cholesterol is not the only end product of the cholesterol synthesis pathway that is inhibited at the stage of mevalonate production by statins[11]
(see ‘Figure 1: Metabolic pathways and products potentially affected by statins’).
Mitochondria play important roles in cellular energy provision and metabolism through the tricarboxylic acid (TCA) cycle, oxidative phosphorylation, and beta-oxidation of fatty acids, cell division, production of reactive oxygen species, and programmed cell death (apoptosis)[13]
. Mitochondrial dysfunction is believed to be the main mechanism of statin-induced myopathy[13],
[14]
. Mitochondria are ubiquitously present in human body cells and it is not surprising that statins can cause side effects related to other body organs, such as the liver, brain, kidney and pancreas[15],
[16]
. It has been suggested that statins can play an antagonistic role on the conductance of chloride channels of muscular cells and they may induce a chronic muscle contractile state, eventually leading to myopathy, in conditions of genetic predisposition, or after efforts by the muscles[17]
.
Statins act by reducing both the flux through the cholesterol synthesis pathway and also the production of ubiquinone (co-enzyme Q10), when measured in plasma or leukocytes, therefore potentially affecting mitochondrial function in muscle[18]
. This is considered as one of the possible mechanisms of the muscular side effects associated with statins, however the exact mechanism of statin-related myopathy is not fully understood[5],
[6]
. Recently, a target for statins within the mitochondrion was identified within complex III of the respiratory electron transport chain (ETC) as a co-enzyme Q10 reductase[19]
. The activity of this enzyme can be inhibited by statins by up to 84% in vitro and 18% in vivo
[19]
. The inhibition of complex III, and therefore, the reduction of the respiratory capacity of myocytes, is suggested as a potential mechanism of statin-induced myopathy. Co-enzyme Q10 is a component of mitochondria acting to facilitate electron transport for the production of adenosine triphosphate (ATP)[20]
and statins inhibit mevalonate production, which is a precursor for the formation of co-enzyme Q10. Co-enzyme Q10 can be measured in the plasma, red blood cells, and inside mitochondria, by using magnetic resonance spectroscopy on muscle biopsies. Observational and randomised controlled trials (RCTs) have demonstrated that statins may reduce circulatory concentrations of co-enzyme Q10 by between 16% and 54%[20]
. Co-enzyme Q10 is largely bound to low-density lipoprotein (LDL) in the circulation, and reduction in LDL concentrations post-statin treatment may also be a contributory factor to its reduced circulatory levels. Reduced plasma and erythrocyte levels of co-enzyme Q10, as well as its depletion in muscle biopsies of animal models and humans, have been reported[20]
. Co-enzyme Q10 depletion results in mitochondrial dysfunction, cell energy reduction, the activation of cell apoptosis pathways, promotion of oxidative stress, and the unravelling of silent mitochondrial defects, which are regarded as possible mechanisms of myopathies associated with statin use[21]
.
Farnesyl and geranyl derivatives, which act to prenylate proteins, are other products of the cholesterol synthesis pathway and have pivotal roles in the biosynthesis of muscle proteins, intracellular signalling, control of cell apoptosis, and gene expression[11]
. Impairment of the synthesis of prenylated proteins and changes in calcium homoeostasis may also be a contributory mechanism of the muscular side effects of statins[12]
. It should be emphasised, however, that few such mechanisms have been confirmed to be the root cause of any of the statin side effects described.
All tissues and organs rely on a balance between cell growth and apoptosis. In vitro studies have demonstrated that statins can induce apoptosis in many cell lines, including skeletal muscle[22]
. Mitochondria damaged by statins can release cytochrome C, and other proteins, leading to the activation of endoproteases, or caspases, and eventually cell death[23],
[24]
. The production of prenylated proteins, which have an essential role in the control of cell growth and apoptosis, are impaired by statins, resulting in increased myocyte cell apoptosis and myopathy (see ‘Figure 1: Metabolic pathways and products potentially affected by statins’). In addition, Heme-A, a ferrous iron porphyrin compound within the ETC, is an essential component of the oxidative phosphorylation pathway and cell respiration[25]
. Its production is also inhibited by statins, likely promoting mitochondrial damage and myopathy. Morphological studies have demonstrated that statins can induce pathological skeletal muscle changes in, essentially, all treated patients[26]
. However, recent studies have found that no significant morphological changes are evident, but the data remain contradictory[27]
.
Myopathy has also been reported in association with other lipid-lowering agents, such as niacin, fibrates and ezetimibe, although much less frequently than with statins. This may indicate that reduced intramuscular cholesterol concentration can, by itself, be a contributory factor to myopathy, induced by statins rather than by a direct toxic effect[24],
[26]
.
Epidemiology of statin-related adverse events
RCTs of statins report low rates of treatment-emergent adverse events both in phase III studies[4]
and then later in CVD outcome trials[28],
[29]
, with minimal numbers of cases of myositis or rhabdomyolysis identified[7]
. However, clinical practice is different. Cohort and registry database studies report a high rate of poor adherence to statin therapy among patients at high risk for CVD, resulting in suboptimal control and management of this condition[30],
[31]
. A study comparing long-term use of evidence-based therapies for CVD demonstrated a rate of 44% compliance for statins compared with 71% for aspirin and 46% for beta blockers[32]
. The Understanding Statin Use in America and Gaps in Patient Education (USAGE) study reports that 67% of patients discontinued statins because of their side effects[31]
. Non-trial studies report variable rates of statin discontinuation from 27% to 77% in primary and secondary prevention of CVD, with the highest rate occurring in primary prevention settings, and within the first two years of starting treatment[30]
. Statin side effects, especially muscle-related ones, are the main reasons given for non-adherence or discontinuation of the treatment[6]
. The spectrum of statin side effects is summarised in ‘Table 1: Summary of statin-related side effects’.
| Muscle-related side effects | Non-muscle-related side effects |
|---|---|
| Symptomatic myopathy | Hepatotoxicity |
| Asymptomatic myopathy | Diabetes mellitus (DM) |
| Myositis (including autoimmune necrotising myositis) | Renal insufficiency and proteinuria |
| Rhabdomyolysis | Neurocognitive and neurological impairments |
| Intracranial haemorrhage (ICH) and depression | |
| Interstitial lung disease (ILD) | |
| Pancreatitis | |
| Lupus-like reaction | |
| Intracranial haemorrhage (ICH) | |
| Sleep disturbance, headache, dizziness, fatigue, depression, sexual dysfunction, etc. |
Muscle-related statin side effects
Statin-induced myopathy ranges from clinically asymptomatic muscle disease recognised by elevated serum creatine kinase (CK) activity to myalgia and life-threatening rhabdomyolysis[5]
. A European consensus panel has classified statin-related myopathy into six grades based on clinical symptoms and the extent of CK changes (see ‘Table 2: A consensus group standardised classification of statin-related side effects and their frequency’ for the classification)[33]
. The incidence of myopathy associated with statins is largely related to the dose and type of statin in use, age, gender, body mass index (BMI), genetics, ethnicity, other comorbidities, and concurrent medications[34],
[35]
. In contrast to biochemically-defined rhabdomyolysis (CK >10 times the upper limit of normal (ULN); 2,500 IU/L), myalgia associated with statins has rarely been systematically studied in clinical trials because of its objective and immeasurable nature[35]
. Guideline groups[6],
[9]
, and the NLA Muscle Safety Expert Panel[10]
, suggested that these side effects can occur with all statins and with varying severity between individuals. Data from the Food and Drug Administration (FDA) Adverse Event Reporting System (AERS) report the incidence of fatal rhabdomyolysis at less than 1 per 1 million prescriptions for all forms of statins as of June 2001[36]
, except for cerivastatin, which was withdrawn from the market in August 2003 because of its high adverse event rate allied with drug interactions[37]
.
| Statin-related myopathy grade | Phenotype | Incidence | Definition |
|---|---|---|---|
| CK: Creatine kinase, ULN: Upper Limit of Normal, Anti-HMGCR: anti-HMG-CoA reductase. Adapted from Alfirevic et al with permission[33] | |||
| 0 | CK rise | 1–26% | Asymptomatic |
| 1 | Myalgia (mild) | 190/105 patient years 0.3–33% | Muscle ache but no CK rise |
| 2 | Myalgia (severe) | 0.2–2 per 103 | Muscle ache; CK 4 × ULN, resolves |
| 3 | Myopathy (mild) | 5–105 patient years | Muscle ache; CK 4–10 × ULN, resolves |
| 4 | Myopathy (severe) | 0.11% | Muscle ache; CK 10–50 × ULN, resolves |
| 5 | Rhabdomyolysis | 0.1–(8.4)/105 patient years | CK 50 × ULN or CK > 10 × ULN with acute kidney injury |
| 6 | Autoimmune necrotising myositis | 2/106 per year | Anti-HMGCR Ab (+) Non-resolving |
Myopathy
The definition of ‘statin-induced myopathy’ varies among studies and experts, resulting in a variable incidence of myopathy associated with statins[6],
[35]
. ‘Myopathy’ is used as a general term to describe clinical or biochemical muscular disturbance[10],
[35]
. The NLA Muscle Safety Expert Panel suggests classifying myopathy as symptomatic myopathy, myositis, asymptomatic myopathy, and rhabdomyolysis (clinically-significant myopathy)[35]
. There are conflicting reports regarding the incidence of myopathy associated with statins, ranging from 1.5–5% in RCTs to 5–20% in observational studies[5]
. Moreover, data from clinical experience and patient registries report an even higher rate of 7–29% of muscle problems induced by statins[6]
. However, the real rate is probably lower as many patients exhibit symptoms with placebo or other lipid-lowering agents but tolerate re-challenge both in blinded and open phases of trials[26]
.
The ODYSSEY alternative study, published in 2015, randomised 361 patients with a history of intolerance to two or more statins, to non-statin methods of LDL reduction (alirocumab, a proprotein convertase subtilisin kexin-9 (PCSK-9) inhibitor, or ezetimibe), and re-challenge with atorvastatin 20mg, or placebo[26]
. The study also had a placebo run-in phase. In the study, 13% discontinued during the placebo run-in phase with 50% attributing it to muscle-related side effects (19% myalgia and 15% muscle spasms). During the 24-week blinded therapy phase, 25–35% discontinued treatment, again, mainly because of myalgia. Rates of discontinuation were 18% for alirocumab and 25% for either ezetimibe or atorvastatin therapy. However, once in the open-label phase of study, 90% persisted with statin therapy if previously assigned to that group. Therefore, much myalgia associated with statins is highly likely to represent pre-existing osteological or neurological complaints and not to be related to treatment with these agents.
In people with recurrent muscle pain on statin use, myopathy can persist for up to two years after discontinuation of treatment[38]
. Statins can uncover clinically silent musculoskeletal conditions such as autoimmune muscle disease (often previously sub-clinical polymyalgia rheumatica), or primary muscle disease including McArdle’s disease, acid maltase deficiency, myoadenylate deaminase deficiency, muscular dystrophy, or even acute intermittent porphyria. All of these can be the reason for persistent myopathy after stopping statin treatment[38],
[39]
.
Symptomatic myopathy
Muscle aches not accompanied by raised CK levels[5],
[12]
define symptomatic myopathy. It refers to muscular symptoms, such as pain, ache and soreness, weakness (which can be confirmed by physical examination), and cramps. These symptoms may occur at any time during the course of treatment, but are usually seen within the first four to six weeks of commencing statin therapy[34]
. It generally affects large proximal muscles, such as gluteus, calf, thigh, and back, and symptoms are usually symmetrical. The reported incidence rate of muscle complaints (myalgia) varies widely from 1% in RCTs to 25% in observational studies[5],
[12]
. This discrepancy could be explained by selection of patients in terms of comorbidities, co-prescribed medications, or to avoid potential problems, and it also could be a result of randomised (or cohort) trials not systematically evaluating these symptoms[40]
. However, a genuine syndrome does exist as evidence of mitochondrial dysfunction, intracellular lipid accumulation, reduced cytochrome oxidase activity, and pathologic changes in muscular fibres, have been shown in muscle biopsies of a small group of patients with statin-associated muscle weakness (objective and subjective) and normal CK concentrations[41],
[42]
.
Asymptomatic myopathy
The NLA Muscle Safety Expert Panel has suggested asymptomatic myopathy should refer to CK elevation without any sign or symptom of myopathy[35]
. Patients may remain asymptomatic with CK levels up to ten times greater than the ULN; this is a common finding in some patients from West Africa[38]
. The NLA Muscle Safety Expert Panel suggested that any increase of CK concentrations is, by strict definition, an indicator of muscle cell breakdown (i.e. rhabdomyolysis)[7]
. The extent of CK rise required to help diagnose myopathy is poorly defined but one study suggests a >140% rise from baseline is likely suspicious[43]
. However, routine measurement of serum CK levels prior to the initiation of statin therapy in patients with no symptoms suggestive of muscular diseases is not recommended because findings are unlikely to impact on clinical patient management and outcomes[2],
[6],
[12]
.
Guidelines recommend that statin treatment should be stopped immediately if CK values are found to be five or more times ULN, or rise significantly to above 1,000 IU/L (which is often used as the acceptable cut-off) from baseline levels[2],
[6]
.
Myositis
Myositis refers to a combination of acute muscle ache or pain, or other muscular complaints, allied with the presence of increased CK levels[5],
[6],
[12]
. Myositis can be caused by factors such as extreme exercise[44]
and can be exacerbated by statin or other drug therapies[45]
. Inflammatory cell infiltration within the skeletal muscle appears to be a natural healing response secondary to the muscular damage[24]
.
A necrotising autoimmune myopathy (NAM) associated with statin treatment, which can be identified by the presence of anti-HMG-CoA reductase (anti-HMGCR) antibodies in serum[46],
[47]
, has recently been identified. Histologically, macrophages are the prominent inflammatory cells in this type of myopathy, making it distinguishable from other forms of autoimmune myositis, such as dermatomyositis (DM) and polymyositis (PM) in which the inflammatory cells infiltrate is composed primarily of lymphocytes[48]
. The damage to the muscle is permanent and characterised by degenerative and regenerative changes of muscular fibres on biopsy. This recently-defined entity can be the cause of permanent muscle weakness associated with statin therapy that is reported by some patients[42]
. A large cohort study reported the presence of anti-HMGCR antibody in patients with background autoimmune myopathies, such as PM and DM, who have never been exposed to statins. Statins can cause or unmask other autoimmune myopathies[49],
[50]
.
Rhabdomyolysis
Significant myopathy is rare and has an incidence of less than 0.5%; however, progressive muscle symptoms can result in fatalities if overlooked[7]
. Rhabdomyolysis is the most severe and potentially life-threatening adverse effect of statins. It can result in renal failure, disseminated intravascular coagulation, multi-organ failure, and death[2],
[6],
[12]
. Statin-induced rhabdomyolysis has been defined and approached differently in research and clinical practice. Rhabdomyolysis refers to severe muscle damage with high CK concentrations, variably defined as more than 10 times to more than 50 times the ULN (usually 150–200 IU/L) with or without renal impairment (e.g. presence of acute kidney injury and/or myoglobunuria). The NLA Muscle Safety Expert Panel suggests redefining statin-induced myopathy terminology in order to make a more practical comparison between different research studies and improve patient care. The panel suggests using classes of CK elevations to replace the rhabdomyolysis term as follows:
- Mild CK increase, defined by rise in CK levels but <10 times the ULN;
- Moderate CK increase, defined by rise in CK levels ≥10 times the ULN but <50 times the ULN;
- Severe CK increase, defined by rise in CK levels ≥50 times the ULN[7],
[23]
.
The NLA Muscle Safety Expert Panel emphasises that pure elevation of CK without symptomatic muscle pain, even if severe, is not a predictor of poor patient outcomes[7]
.
Genetic risk factors for myopathy associated with statins
Statins are lactone pro-drugs metabolised in the liver to active statin acids. The risk of myopathy rises with increased statin dose. Statins are either lactone prodrugs (e.g. simvastatin, lovastatin) or acids that are metabolised to unstable derivatives by glucuronosyl transferase enzymes, which then decompose to give statin lactones that are substrates for the cytochrome P450 (CYP) 3A4 system[51]
. Most commonly used statins are metabolised by, or bind to, CYP3A4. Drug metabolism studies have demonstrated that simvastatin and lovastatin blood levels are more likely to be affected by cytochrome P450 system inhibitors, while atorvastatin is less affected by this pathway. Pravastatin and rosuvastatin do not inhibit CYP3A4 and so are less prone to interference from drugs that also inhibit this pathway (e.g. some antibiotics, such as clarithromycin, systemic ‘conazole’ antifungals, and some HIV protease inhibitors). Other medications, including calcium channel blockers (e.g. verapamil, and especially diltiazem), amiodarone, corticosteroids, antipsychotics, colchicine, digoxin, and warfarin, can increase risk of statin toxicity[5],
[12]
. Excessive consumption of alcohol, pomegranate, cranberry and grapefruit juice (also CYP3A4 inhibitors), and other factors such as drug abuse (cocaine, amphetamines, heroin), previous statin-induced myotoxicity, heavy exercise, female gender, being of Asian descent, advanced age (>65 years), low BMI, renal and hepatic impairment, including biliary tree obstruction, hypothyroidism, vitamin D deficiency and background muscular metabolic disorders, are all associated with increased risk of myopathy in statin treatment[5],
[6],
[12]
.
Some studies have tried to identify genetic risk factors for statin-associated myopathy[52]
. The only consistent association was found in the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) trial. A genome-wide association study showed a strong association between myopathy caused by high-dose simvastatin therapy (80mg) and a single-nucleotide polymorphism (SNP) located within the SLCO1B1 gene on chromosome 12 encoding organic anion transporting polypeptide (OATP) (P <0.001)[53]
. The SLCO1B1 gene has been shown to regulate the hepatic uptake of statin acids and the C allele variant of this gene has been observed to be associated with 60% of cases of statin-associated myopathy in some series. It has been suggested that the variant C polymorphism of SLCO1B1 is likely to affect the plasma concentrations of all statins but few studies have confirmed this[54]
.
Statin acids are excreted following a glucuronidation process performed by UDP-glucuronosyltransferase 1A1 (UGT1A1). This was identified as the source of myositis that was induced by an interaction between cerivastatin and gemifibrozil[55]
. Both high-dose nicotinic acid (Niacin) and fibric acid derivatives can increase the risk of myopathy and rhabdomyolysis when co-prescribed with statins[24]
. Case reports also suggest ezetimibe per se, or especially when combined with a statin, may increase the risk of myopathy. The mechanism of ezetimibe-induced myopathy is not clear[56]
.
Prevention and management of muscle-related adverse effects of statins
Current guidelines recommend high-dose, high-intensity statin treatment in all patients admitted for CVD regardless of their cholesterol concentration, and the initiation of moderate–dose, high-intensity statin therapy in all high-risk patients for primary prevention[2]
. The chance of statin withdrawal in primary care settings is high and associated with increased long-term CVD mortality[30]
. Therefore, educating patients about the role and side effects of these drugs is essential to promoting adherence. Causes of non-adherence with statins are complex and are mainly a result of patients’ concerns about, or experiences with, adverse effects, and especially myopathy[31],
[32]
.
Measures can be taken to reduce the risk of myopathy and therefore improve adherence to statins[57]
. A full clinical assessment, as well as obtaining a thorough medical history, can help identify patients at risk of statin-induced myopathy or other known adverse effects[6],
[12]
. Hypothyroidism is associated with elevated cholesterol levels and an increased risk of myopathy with statins[58]
. Any significant thyroid dysfunction should be corrected prior to the initiation of statin therapy. Vitamin D deficiency is associated with myalgia and may be associated with increased rates of statin-induced myopathy[59]
. Some centres recommend treatment of gross vitamin D deficiency prior to initiation of statin therapy, but there are few studies to support this idea. There is no convincing evidence to support that co-enzyme Q10[60]
, magnesium, vitamin D[61]
or vitamin E[60]
supplementation can reduce the risk of myopathy associated with statins.
Hydrophilic statins are thought to be more actively transported into hepatocytes through expressing OATP, and less likely to diffuse into muscles or other peripheral cells as seen with lipophilic derivatives[62]
. Lovastatin and simvastatin are the most lipophilic ones, followed by atorvastatin, fluvastatin, rosuvastatin and pravastatin. Guidelines suggest the primary use of atorvastatin given its superior efficacy and safety profile compared with simvastatin[4],
[63]
. Prescribing a less lipophilic statin, initiating an intermittent statin regimen, or employing a low statin dose combined with non-statin lipid-lowering therapies in high-risk individuals are all used to reduce the chance of inducing side effects.
If patients develop muscle-related symptoms when on statin therapy, CK levels should be measured and if >10 times the ULN, statins should be immediately discontinued. If there is any suggestion of significant dehydration, prompting re-hydration to prevent renal failure is necessary. The UK National Institute for Health and Care Excellence (NICE) guidelines suggest that in the absence of muscle symptoms, statins can be continued if CK values are <5 times the ULN[2]
. The presence of other causes of myopathy, such as concurrent hypothyroidism and use of CYP3A4 inhibitors, should be excluded in patients developing muscle symptoms while on statins. Plant stanols, alongside dietary and lifestyle modifications, can be introduced to low-dose statin treatment regimens as complementary measures.
A proposed algorithm for the management of muscular side effects of statins is provided in ‘Figure 2: Proposed algorithm for the management of muscular side effects of statins prior to, and after starting, treatment’.
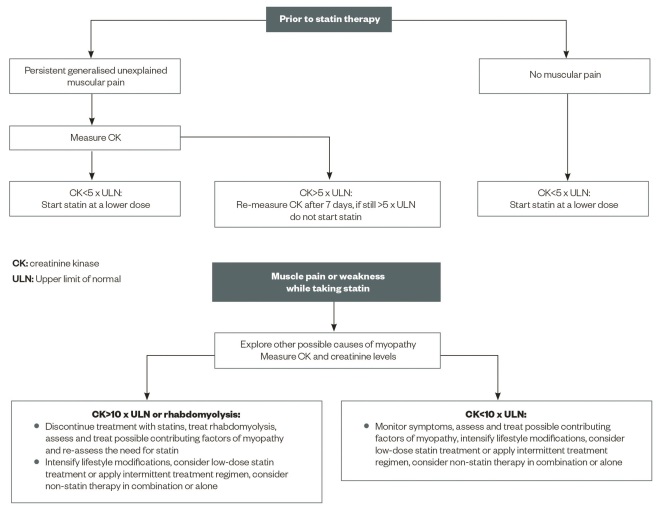
Figure 2: Proposed algorithm for the management of muscular side effects of statins prior to, and after starting, treatment
A clinical scheme for dealing with patients complaining of myopathy that may be related to statin therapy
Statin therapy should be amended to lower the risk of myopathy, such as using reduced doses, or weaker statins, such as fluvastatin or pravastatin, or using intermittent dosing of long-acting forms, such as rosuvastatin (used 1–3 times a week)[6],
[12]
. Red yeast rice preparations contain variable quantities of a statin homologue, mevinolin/lovastatin, and have been used successfully in some individuals[64]
. If a patient tolerates red yeast rice, efforts should be made to reinstitute pharmacological statin therapy[65]
.
In secondary prevention, statin monotherapy using these approaches is often inadequate and non-statin treatments such as ezetimibe, or less commonly bile acid sequestrants, can be added. If patients prove intolerant to these combinations, then monotherapy with ezetimibe, fibrates, or in some cases niacin, may be appropriate. The newest alternative therapy for statin-intolerant patients with severe dyslipidaemia or CVD is the prescription of PCSK-9 inhibitors[26]
. These are, however, expensive and therefore their use is currently restricted. In extreme cases, LDL apheresis can be used in the treatment regimen of dyslipidaemia but it is only of limited efficacy in the absence of between-procedure lipid-lowering therapy. If all else fails, there is evidence that ileal bypass surgery can reduce LDL-cholesterol (LDL-C) and reduce CVD events, but few centres have the requisite expertise to perform this procedure[66]
.
Non-muscle-related statin side effects
Statins are associated with some common side effects that vary between different forms of statins. Some clinical trials have revealed that some statin-induced side effects are not statistically different from placebo[4],
[7]
. The reported side effects associated with statins commonly include gastrointestinal disturbance and asymptomatic increases in hepatic transaminases, which may affect up to one in ten statin users. More recently, a consistent association between initiation of statin therapy and the exacerbation of dysglycaemia, sometimes leading to the reclassification of individuals to a diagnosis of type 2 diabetes, has been described. Rarer side effects include sleep disturbance, headache, dizziness, depression, paraesthesiae, asthenia, amnesia, fatigue, sexual dysfunction, breast enlargement, thrombocytopenia, arthralgia, visual disturbance, alopecia, rash, pruritus, urticarial, and non-allergic rhinitis. In very rare cases, statins can cause more serious conditions, such as pancreatitis and interstitial lung diseases, or a Lupus-like reaction.
Hepatotoxicity
Statins are associated with a broad spectrum of hepatic adverse effects commonly including an asymptomatic rise in plasma transaminase concentrations[4],
[67]
. Use of statins in active liver disease is usually contraindicated[67]
. Transaminitis is a benign condition, is usually transient, and often occurs on initiation of the drug[68]
. It is not associated with any histological changes in hepatic tissue. The pathophysiology of this rise is unclear and is thought to be caused by changes in the hepatocellular lipid concentration and plasma membrane permeability, which may cause transaminase enzyme leak and an associated inflammatory response giving rise to a ‘biochemical hepatitis’[69]
. The NLA Liver Expert Panel confirms that elevations in serum transaminases can occur with all forms of statins currently available and with all doses[70]
. However, elevated serum transaminase concentrations >3 times ULN are seen in <1% patients on monotherapy with statins, except for simvastatin 80mg, which is associated with a 2–3% increased risk[71]
. It is difficult to establish a causal relationship between statin use and aminotransferase elevation on account of a high concurrent rate of non-alcoholic fatty liver disease (NAFLD) in hyperlipidaemic patients. A post-hoc analysis of the Greek Atorvastatin Clinical Evaluation (GREACE) study has suggested that in patients with mild transaminitis and likely with NAFLD, statins may improve the degree of hepatic dysfunction[72]
. The NLA Liver Expert Panel suggested that routine monitoring of liver function tests was not necessary in patients receiving statins and the FDA has removed this requirement from the prescribing information for statins. However, most countries still say annual transaminase monitoring for patients on statin therapy is necessary.
Rare case reports of liver failure associated with statin use appear to be an idiosyncratic reaction. Patients who develop a severe rise in serum transaminase concentrations >10 times ULN during treatment with statins usually have other background comorbidities and use concurrent medications that potentially interfere with statin metabolism in the liver. UK NICE guidelines recommend the discontinuation of statins in cases of persistent elevated transaminases levels >3 times ULN (>150 IU/L), but also suggest that minor elevations in transaminases should not preclude initiation of statin therapy[2]
.
Diabetes mellitus
A meta-analysis of RCTs showed that statins are associated with 9% increased risk for type 2 diabetes mellitus (DM) compared with placebo or standard care (odds ratio [OR]: 1.09; 95% CI: 1.02–1.17)[73]
. This finding was followed by a FDA alert on statin safety in 2012 and observational studies that demonstrated similar outcomes. A population-based primary care cohort study suggested that treatment with higher potency statins, especially atorvastatin and simvastatin, may be associated with an increased risk of new-onset diabetes (atorvastatin, adjusted hazard ratio [HR] 1.22; 95% CI: 1.15–1.29; rosuvastatin, HR 1.18, CI: 1.10–1.26; and simvastatin, HR 1.10, CI: 1.04–1.17)[74]
. A further meta-analysis of five clinical trials suggested that the effect may be dose-dependent because high-dose statin treatment is associated with new-onset diabetes compared with moderate-dose statin therapy (OR 1.12; 95% CI: 1.04–1.22)[75]
. This effect seems to be real and to be associated with weight gain secondary to statin therapy as shown by a Mendelian randomisation study of functional HMG-CoA reductase polymorphisms that was also associated with a greater incidence of diabetes[76]
. Nevertheless, the benefits of statin therapy for CVD events significantly outweigh the slight increased risk of a diagnosis of type 2 DM[76]
.
Renal insufficiency and proteinuria
There is no evidence that statins are associated with renal impairment or are a cause of acute renal failure (ARF) in the absence of rhabdomyolysis[77]
. The reports of acute interstitial nephritis in association with statins might be caused by an allergic reaction in the interstitial tissue of the kidney and not because of a direct nephrotoxic effect. A meta-analysis of large RCTs did not find an increased incidence of acute renal failure following statin therapy[4]
. The NLA Renal Expert Panel does not recommend routine monitoring of renal function in patients receiving statins and other guidelines have followed suit. Some case reports have identified proteinuria with statins. In an early phase III clinical trial of rosuvastatin at an 80mg dose (the currently approved dose range is 5–40mg), an increase in urine protein of ≥ 2+ on a dipstick compared with placebo, and other types of statins, was reported[77],
[78]
. The NLA Renal Expert Panel was not convinced that statins are associated with proteinuria and recommended the use of more potent statins in patients who have an increased albumin/creatinine ratio as part of their CVD risk profile. Statins may have beneficial effects on albuminuria, an independent risk factor for CVD and renal diseases[79]
, as there seems to be some effect of statin therapy in reducing progression of renal function deterioration, demonstrated in patients with established renal disease[80]
.
Neurocognitive and neurological impairments
In 2012, the FDA, in a consumer and healthcare professional advisory capacity, raised concerns regarding the risk of cognitive impairment, such as memory loss, forgetfulness, and confusion, in statin users based on data from the Adverse Events Reporting System (AERS) and medical literature[81],
[82]
. Case report and case-series studies are controversial. One retrospective case-control study supports a strong correlation between dose-dependent statin therapy and acute memory loss[83]
. Placebo-controlled RCTs and a meta-analysis of cognitive outcome data failed to show any association between statin therapy and cognitive decline in patients with, or without, background cognitive impairment, such as those with Alzheimer’s disease[81]
. However, the conflicting data may be a consequence of different statins being studied (i.e. their lipophilicity), the dose and duration of statin in use, the choice of control group, and the limited sample size in some studies.
The NLA Neurology Expert Panel, in 2006, found no evidence to support the suggestion that statins increase the risk of peripheral neuropathy, as no association was found in the Heart Protection Study (HPS) or the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) studies[82]
. The small number of epidemiologic, or case, studies that have reported correlations between statin use and peripheral neuropathy[84]
could be biased by the fact that patients likely to receive statin treatment were already at increased risk for peripheral neuropathy by having comorbidities such as DM, insulin resistance, and advanced vascular diseases[85],
[86],
[87]
.
Intracranial haemorrhage and depression
Lipoproteins are involved in providing surfaces for the binding of coagulation factor, and anticoagulant proteins are associated with high-density lipoproteins (HDL). Epidemiological studies have suggested that low LDL-C is associated with increased rates of cerebral haemorrhage. However, a meta-analysis of 31 RCTs of statin therapy reported no significant correlations between active statin use and an increased risk of intracranial haemorrhage (ICH)[88]
. A retrospective cohort study of 3481 hospitalised patients with ICH showed that statin-users have a better clinical outcome and a higher post-event survival rate compared with non-users[89]
. It has also been suggested that statins are associated with increased risks of depression as a consequence of reduced lipid concentrations. However, clinical conditions, such as myocardial infarction and stroke, are associated with depression by themselves[82]
.
Interstitial lung disease
One rare, yet serious, side effect of statins[56],
[57]
is interstitial lung disease (ILD). Several cases of statin-induced ILD have been reported in the literature, mostly associated with simvastatin, fluvastatin, and atorvastatin[90],
[91]
. A literature review described similar, and non-diagnostic, pathologic findings such as inflammatory cell infiltration, and fibrosis of pulmonary parenchyma, endothelial basement membranes and pleura in patients, developing as pulmonary adverse effects to statins[91]
. It has been suggested that the possible mechanisms of ILD associated with statins include humoral and cell-mediated immune responses, as well as mitochondrial impairment and the inhibition of phospholipase production as part of the metabolic effects of statins.
Pancreatitis
Systematic reviews and case report studies have shown that pancreatitis can occur at any stage of treatment with statins and at both low and high doses[92],
[93]
. The mechanism for statin-induced pancreatitis remains ill-defined. A mechanism involving an immune-mediated inflammatory response and a direct cellular toxicity effect have been proposed, but statins are generally anti-inflammatory through their action on protein prenylation and rho kinase A. In 2012, a meta-analysis of clinical trials with statins suggests they may decrease rates of pancreatitis through their anti-inflammatory action[94]
.
Lupus-like reaction
A lupus-like reaction, although very rare, is a reported side effect of long-term use of statin therapy. It is characterised by an auto-immune inflammatory response and presence of anti-dsDNA antibodies in the circulation[95]
. Statins are known to induce cell apoptosis, which may result in the exposure of cell nucleus antigen to the immune system, subsequently causing an inflammatory response[96]
. Simvastatin and atorvastatin, in particular, are potent pro-apoptotic agents and are thought to be more related to lupus-like syndrome and other skin autoimmune conditions, such as dermatomyositis and lichen planus pemphigoides. Nevertheless, establishing a causal relationship between statin use and an autoimmune inflammatory response appears to be difficult on account of asynchrony between statin initiation and autoimmunity on the one hand, and discontinuation of the treatment and disappearance of the inflammatory response on the other.
Conclusion
Statins are the only agents that have been proved to be both clinically efficient and financially affordable in the treatment or management of patients at high risk of CVD. Several studies have confirmed that the cardiovascular benefits of statin treatment in high-risk populations outweigh the rare adverse effects, such as rhabdomyolysis. Clinicians are recommended to take every effort to keep such patients on statin-based lipid-lowering strategies and should not be fearful of mild muscular symptoms or insignificant elevations of CK. Patients must be fully educated and thoroughly informed about the benefits and risks of statin treatment to maintain long-term adherence.
Negar Maghsoodi is a specialist registrar in the Department of Metabolic Medicine/Chemical Pathology, Guy’s and St Thomas’ Hospitals, London, and Anthony S Wierzbicki is consultant (honorary professor) in metabolic medicine/chemical pathology at the Department of Chemical Pathology, Guy’s and St Thomas’ Hospitals, London. Correspondence to:
anthony.wierzbicki@kcl.ac.uk
Financial and conflicts of interest disclosure:
The authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilised in the production of this manuscript.
Reading this article counts towards your CPD
You can use the following forms to record your learning and action points from this article from Pharmaceutical Journal Publications.
Your CPD module results are stored against your account here at The Pharmaceutical Journal. You must be registered and logged into the site to do this. To review your module results, go to the ‘My Account’ tab and then ‘My CPD’.
Any training, learning or development activities that you undertake for CPD can also be recorded as evidence as part of your RPS Faculty practice-based portfolio when preparing for Faculty membership. To start your RPS Faculty journey today, access the portfolio and tools at www.rpharms.com/Faculty
If your learning was planned in advance, please click:
If your learning was spontaneous, please click:
References
[1] Stone NJ, Robinson JG, Lichtenstein AH et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129:S1–S45. doi: 10.1161/01.cir.0000437738.63853.7a
[2] Rabar S, Harker M, O’Flynn N et al. Lipid modification and cardiovascular risk assessment for the primary and secondary prevention of cardiovascular disease: summary of updated NICE guidance. BMJ 2014;349:g4356. doi: 10.1136/bmj.g4356
[3] Alsheikh-Ali AA, Maddukuri PV, Han H et al. Effect of the magnitude of lipid lowering on risk of elevated liver enzymes, rhabdomyolysis, and cancer: insights from large randomized statin trials. J Am Coll Cardiol 2007;50:409–418. doi: 10.1016/j.jacc.2007.02.073
[4] Naci H, Brugts J & Ades T. Comparative tolerability and harms of individual statins: a study-level network meta-analysis of 246 955 participants from 135 randomized, controlled trials. Circulation Cardiovascular Quality and Outcomes 2013;6:390–399. doi: 10.1161/CIRCOUTCOMES.111.000071
[5] Joy TR & Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009;150:858–868. doi: 10.7326/0003-4819-150-12-200906160-00009
[6] Stroes ES, Thompson PD, Corsini A et al. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J 2015;36:1012–1022. doi: 10.1093/eurheartj/ehv043
[7] Armitage J. The safety of statins in clinical practice. Lancet 2007;370:1781–1790. doi: 10.1016/S0140-6736(07)60716-8
[8] Mancini GB, Baker S, Bergeron J et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: proceedings of a Canadian Working Group Consensus Conference. Can J Cardiol 2011;27:635–662. doi: 10.1016/j.cjca.2011.05.007
[9] Mancini GB, Tashakkor AY, Baker S et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol 2013;29:1553–1568. doi: 10.1016/j.cjca.2013.09.023
[10] McKenney JM, Davidson MH, Jacobson TA et al. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006;97:89C–94C. doi: 10.1016/j.amjcard.2006.02.030
[11] Wierzbicki AS, Poston R & Ferro A. The lipid and non-lipid effects of statins. Pharmacol Ther 2003;99:95–112. doi: 10.1016/S0163-7258(03)00055-X
[12] Stock J. Statin-associated muscle symptoms EAS Consensus Panel paper focuses on this neglected patient group. Atherosclerosis 2015;242:346–350. doi: 10.1016/j.atherosclerosis.2015.06.049
[13] Johnson TE, Zhang X, Bleicher KB et al. Statins induce apoptosis in rat and human myotube cultures by inhibiting protein geranylgeranylation but not ubiquinone. Toxicol Appl Pharmacol 2004;200:237–250. doi: 10.1016/j.taap.2004.04.010
[14] Matzno S, Yasuda S, Juman S et al. Statin-induced apoptosis linked with membrane farnesylated Ras small G protein depletion, rather than geranylated Rho protein. J Pharm Pharmacol 2005;57:1475–1484. doi: 10.1211/jpp.57.11.0014
[15] Vaklavas C, Chatzizisis YS, Ziakas A et al. Molecular basis of statin-associated myopathy. Atherosclerosis 2009;202:18–28. doi: 10.1016/j.atherosclerosis.2008.05.021
[16] Golomb BA & Evans MA. Statin adverse effects: a review of the literature and evidence for a mitochondrial mechanism. Am J Cardiovasc Drugs 2008;8:373–418. doi: 10.2165/0129784-200808060-00004
[17] Pierno S, Camerino GM, Cippone V et al. Statins and fenofibrate affect skeletal muscle chloride conductance in rats by differently impairing ClC-1 channel regulation and expression. Br J Pharmacol 2009;156:1206–1215. doi: 10.1111/j.1476-5381.2008.00079.x
[18] Folkers K, Langsjoen P, Willis R et al. Lovastatin decreases coenzyme Q levels in humans. Proc Natl Acad Sci USA 1990;87:8931–8934. PMCID: PMC55074
[19] Schirris TJ, Renkema GH, Ritschel T et al. Statin-Induced Myopathy Is Associated with Mitochondrial Complex III Inhibition. Cell Metab 2015;22:399–407. doi: 10.1016/j.cmet.2015.08.002
[20] Marcoff L & Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007;49:2231–2237. doi: 10.1016/j.jacc.2007.02.049
[21] Deichmann R, Lavie C & Andrews S. Coenzyme q10 and statin-induced mitochondrial dysfunction. Ochsner J 2010;10:16–21. PMID: 21603349
[22] Dirks AJ & Jones KM. Statin-induced apoptosis and skeletal myopathy. Am J Physiol Cell Physiol 2006;291:C1208–C1212. doi: 10.1152/ajpcell.00226.2006
[23] Suzuki Y, Imai Y, Nakayama H et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 2001;8:613–621. doi: 10.1016/S1097-2765(01)00341-0
[24] Cafforio P, Dammacco F, Gernone A et al. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis 2005;26:883–891. doi: 10.1093/carcin/bgi036
[25] Hederstedt L. Heme A biosynthesis. Biochim Biophys Acta 2012;1817:920–927. doi: 10.1016/j.bbabio.2012.03.025
[26] Moriarty PM, Thompson PD, Cannon CP et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol 2015;9:758–769. doi: 10.1016/j.jacl.2015.08.006
[27] Rengo JL, Callahan DM, Savage PD et al. Skeletal muscle ultrastructure and function in statin-tolerant individuals. Muscle Nerve 2016;53:242–251. doi: 10.1002/mus.24722
[28] Baigent C, Keech A, Kearney PM et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. The Lancet 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1
[29] Baigent C, Blackwell L, Emberson J et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. The Lancet 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5
[30] Penning-van Beest FJ, Termorshuizen F, Goettsch WG et al. Adherence to evidence-based statin guidelines reduces the risk of hospitalizations for acute myocardial infarction by 40%: a cohort study. Eur Heart J 2007;28:154–159. doi: 10.1093/eurheartj/ehl391
[31] Cohen JD, Brinton EA, Ito MK et al. Understanding Statin Use in America and Gaps in Patient Education (USAGE): an internet-based survey of 10,138 current and former statin users. J Clin Lipidol 2012;6:208–215. doi: 10.1016/j.jacl.2012.03.003
[32] Maningat P, Gordon BR & Breslow JL. How do we improve patient compliance and adherence to long-term statin therapy? Curr Atheroscler Rep 2013;15:291. doi: 10.1007/s11883-012-0291-7
[33] Alfirevic A, Neely D, Armitage J et al. Phenotype standardization for statin-induced myotoxicity. Clin Pharmacol Ther 2014;96:470–476. doi: 10.1038/clpt.2014.121
[34] Bruckert E, Hayem G, Dejager S et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc Drugs Ther 2005;19:403–414. doi: 10.1007/s10557-005-5686-z
[35] Thompson PD, Clarkson PM & Rosenson RS. An assessment of statin safety by muscle experts. Am J Cardiol 2006;97:69C–76C. doi: 10.1016/j.amjcard.2005.12.013
[36] Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006;97:44C–51C. doi: 10.1016/j.amjcard.2005.12.009
[37] McClure DL, Valuck RJ, Glanz M et al. Systematic review and meta-analysis of clinically relevant adverse events from HMG CoA reductase inhibitor trials worldwide from 1982 to present. Pharmacoepidemiol Drug Saf 2007;16:132–143. doi: 10.1002/pds.1341
[38] Thompson PD, Clarkson P & Karas RH. Statin-associated myopathy. JAMA 2003;289:1681–1690. doi: 10.1001/jama.289.13.1681
[39] Baker SK, Vladutiu GD, Peltier WL et al. Metabolic myopathies discovered during investigations of statin myopathy. Can J Neurol Sci 2008;35:94–97. PMID: 18380285
[40] Fernandez G, Spatz ES, Jablecki C et al. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med 2011;78:393–403. doi: 10.3949/ccjm.78a.10073
[41] Phillips PS, Haas RH, Bannykh S et al. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med 2002;137:581–585. doi: 10.7326/0003-4819-137-7-200210010-00009
[42] Phillips PS & Haas RH. Statin myopathy as a metabolic muscle disease. Expert Rev Cardiovasc Ther 2008;6:971–978. doi: 10.1586/14779072.6.7.971
[43] Wu AH, Smith A & Wians F. Interpretation of creatine kinase and aldolase for statin-induced myopathy: Reliance on serial testing based on biological variation. Clin Chim Acta 2009; 399:109–111. doi: 10.1016/j.cca.2008.09.023
[44] Sinzinger H & O’Grady J. Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscular problems. Br J Clin Pharmacol 2004;57:525–528. doi: 10.1111/j.1365-2125.2003.02044.x
[45] Kearns AK, Bilbie CL, Clarkson PM et al. The creatine kinase response to eccentric exercise with atorvastatin 10 mg or 80 mg. Atherosclerosis 2008;200:121–125. doi: 10.1016/j.atherosclerosis.2007.12.029
[46] Mohassel P & Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve 2013;48:477–483. doi: 10.1002/mus.23854
[47] Babu S & Li Y. Statin induced necrotizing autoimmune myopathy. J Neurol Sci 2015;351:13–17. doi: 10.1016/j.jns.2015.02.042
[48] Ramanathan S, Langguth D, Hardy TA et al. Clinical course and treatment of anti-HMGCR antibody-associated necrotizing autoimmune myopathy. Neurol Neuroimmunol Neuroinflamm 2015;2:e96. doi: 10.1212/NXI.0000000000000096
[49] Padala S & Thompson PD. Statins as a possible cause of inflammatory and necrotizing myopathies. Atherosclerosis 2012;222:15–21. doi: 10.1016/j.atherosclerosis.2011.11.005
[50] Mammen AL. Statin-Associated Autoimmune Myopathy. N Engl J Med 2016;374:664–669. doi: 10.1056/NEJMra1515161
[51] Neuvonen PJ, Niemi M & Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther 2006;80:565–581. doi: 10.1016/j.clpt.2006.09.003
[52] Vladutiu GD, Simmons Z, Isackson PJ et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 2006;34:153–162. PMID: 16671104
[53] Link E, Parish S, Armitage J et al. SLCO1B1 variants and statin-induced myopathy – a genomewide study. N Engl J Med 2008;359:789–799. doi: 10.1056/NEJMoa0801936
[54] Stewart A. SLCO1B1 Polymorphisms and Statin-Induced Myopathy. PLoS Curr 2013;5. doi: 10.1371/currents.eogt.d21e7f0c58463571bb0d9d3a19b82203
[55] Prueksaritanont T, Tang C, Qiu Y et al. Effects of fibrates on metabolism of statins in human hepatocytes. Drug Metab Dispos 2002;30:1280–1287. doi: 10.1124/dmd.30.11.1280
[56] Simard C & Poirier P. Ezetimibe-associated myopathy in monotherapy and in combination with a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Can J Cardiol 2006;22:141–144. doi: 10.1016/S0828-282X(06)70253-7
[57] Eckel RH. Approach to the patient who is intolerant of statin therapy. J Clin Endocrinol Metab 2010;95:2015–2022. doi: 10.1210/jc.2009-2689
[58] Tokinaga K, Oeda T, Suzuki Y et al. HMG-CoA reductase inhibitors (statins) might cause high elevations of creatine phosphokinase (CK) in patients with unnoticed hypothyroidism. EndocrJ 2006;53:401–405. doi: 10.1507/endocrj.K04-144
[59] Michalska-Kasiczak M, Sahebkar A, Mikhailidis DP et al. Analysis of vitamin D levels in patients with and without statin-associated myalgia – A systematic review and meta-analysis of 7 studies with 2420 patients. International Journal of Cardiology 2014;178C:111–116. doi: 10.1016/j.ijcard.2014.10.118
[60] Banach M, Serban C, Sahebkar A et al. Effects of coenzyme Q10 on statin-induced myopathy: a meta-analysis of randomized controlled trials. Mayo Clinic Proceedings 2015;90:24–34. doi: 10.1016/j.mayocp.2014.08.021
[61] Glueck CJ, Budhani SB, Masineni SS et al. Vitamin D deficiency, myositis-myalgia, and reversible statin intolerance. Curr Med Res Opin 2011;27:1683–1690. doi: 10.1185/03007995.2011.598144
[62] Bitzur R, Cohen H, Kamari Y et al. Intolerance to statins: mechanisms and management. Diabetes Care 2013;36 Suppl 2:S325–S330. doi: 10.2337/dcS13-2038
[63] Naci H, Brugts JJ, Fleurence R et al. Dose-comparative effects of different statins on serum lipid levels: a network meta-analysis of 256,827 individuals in 181 randomized controlled trials. European Journal of Preventive Cardiology 2013;20:658–670. doi: 10.1177/2047487313483600
[64] Liu J, Zhang J, Shi Y et al. Chinese red yeast rice (Monascus purpureus) for primary hyperlipidemia: a meta-analysis of randomized controlled trials. Chin Med 2006;1:4. doi: 10.1186/1749-8546-1-4
[65] Halbert SC, French B, Gordon RY et al. Tolerability of red yeast rice (2,400 mg twice daily) versus pravastatin (20 mg twice daily) in patients with previous statin intolerance. Am J Cardiol 2010;105:198–204. doi: 10.1016/j.amjcard.2009.08.672
[66] Buchwald H, Rudser KD, Williams SE et al. Overall mortality, incremental life expectancy, and cause of death at 25 years in the program on the surgical control of the hyperlipidemias. AnnSurg 2010;251:1034–1040. doi: 10.1097/SLA.0b013e3181deb4d0
[67] Anfossi G, Massucco P, Bonomo K et al. Prescription of statins to dyslipidemic patients affected by liver diseases: a subtle balance between risks and benefits. Nutr Metab Cardiovasc Dis 2004;14:215–224. PMID: 15553600
[68] Wierzbicki AS, Lumb PJ, Chik G et al. Fibrinogen response with simvastatin versus atorvastatin in familial hypercholesterolemia. AmJ Cardiol 2001;87:338–340, A9. doi: 10.1016/S0002-9149(00)01371-0
[69] Wierzbicki AS, Lumb PJ, Semra YK et al. Effect of atorvastatin on plasma fibrinogen. The Lancet 1998;351:569–570. PMID: 9492781
[70] Cohen DE, Anania FA & Chalasani N. An assessment of statin safety by hepatologists. Am J Cardiol 2006;97:77C–81C. doi: 10.1016/j.amjcard.2005.12.014
[71] Armitage J, Bowman L, Wallendszus K et al. Intensive lowering of LDL cholesterol with 80mg versus 20mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. The Lancet 2010;376:1658–1669. doi: 10.1016/S0140-6736(10)60310-8
[72] Athyros VG, Tziomalos K, Gossios TD et al. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: a post-hoc analysis. The Lancet 2010;376:1916–1922. doi: 10.1016/S0140-6736(10)61272-X
[73] Sattar N, Preiss D, Murray HM et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. The Lancet 2010;375:735–742. doi: 10.1016/S0140-6736(09)61965-6
[74] Carter AA, Gomes T, Camacho X et al. Risk of incident diabetes among patients treated with statins: population based study. BMJ 2013;346:f2610. doi: 10.1136/bmj.f2610
[75] Preiss D, Seshasai SR, Welsh P et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA 2011;305:2556–2564. doi: 10.1001/jama.2011.860
[76] Swerdlow DI, Preiss D, Kuchenbaecker KB et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. The Lancet 2015;385:351–361. doi: 10.1016/S0140-6736(14)61183-1
[77] Kasiske BL, Wanner C & O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006;97:82C–85C. doi: 10.1016/j.amjcard.2005.12.015
[78] Guthrie RM & Martin DR. The safety of rosuvastatin: effects on renal and hepatic function. Expert Opin Drug Saf 2007;6:573–581. doi: 10.1517/14740338.6.5.573
[79] Douglas K, O’Malley PG & Jackson JL. Meta-analysis: the effect of statins on albuminuria. Annals of Internal Medicine 2006;145:117–124. doi: 10.7326/0003-4819-145-2-200607180-00009
[80] Athyros VG, Katsiki N, Karagiannis A et al. Statins can improve proteinuria and glomerular filtration rate loss in chronic kidney disease patients, further reducing cardiovascular risk. Fact or fiction? Expert Opin Pharmacother 2015;16:1449–1461. doi: 10.1517/14656566.2015.1053464
[81] Ott BR, Daiello LA, Dahabreh IJ et al. Do Statins Impair Cognition? A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Journal of General Internal Medicine 2015. doi: 10.1007/s11606-014-3115-3
[82] Brass LM, Alberts MJ & Sparks L. An assessment of statin safety by neurologists. Am J Cardiol 2006;97:86C–88C. doi: 10.1016/j.amjcard.2005.12.017
[83] Strom BL, Schinnar R, Karlawish J et al. Statin Therapy and Risk of Acute Memory Impairment. JAMA Intern Med 2015;175:1399–1405. doi: 10.1001/jamainternmed.2015.2092
[84] Gaist D, Jeppesen U, Andersen M et al. Statins and risk of polyneuropathy: a case-control study. Neurology 2002;58:1333–1337. doi: 10.1212/WNL.58.9.1333
[85] Corrao G, Zambon A, Bertu L et al. Lipid lowering drugs prescription and the risk of peripheral neuropathy: an exploratory case-control study using automated databases. J Epidemiol Community Health 2004;58:1047–1051. doi: 10.1136/jech.2003.013409
[86] Gaist D, Garcia Rodriguez LA, Huerta C et al. Are users of lipid-lowering drugs at increased risk of peripheral neuropathy? Eur J Clin Pharmacol 2001;56:931–933. doi: 10.1007/s002280000248
[87] Bhalla S, Singh N & Jaggi AS. Statins: do they aggravate or ameliorate neuropathic pain? J Pain 2014;15:1069–1080. doi: 10.1016/j.jpain.2014.06.012
[88] McKinney JS & Kostis WJ. Statin therapy and the risk of intracerebral hemorrhage: a meta-analysis of 31 randomized controlled trials. Stroke 2012;43:2149–2156. doi: 10.1161/STROKEAHA.112.655894
[89] Flint AC, Conell C, Rao VA et al. Effect of statin use during hospitalization for intracerebral hemorrhage on mortality and discharge disposition. JAMA Neurol 2014;71:1364–1371. doi: 10.1001/jamaneurol.2014.2124
[90] Schwaiblmair M, Behr W, Haeckel T et al. Drug induced interstitial lung disease. Open Respir Med J 2012;6:63–74. doi: 10.2174/1874306401206010063
[91] Fernandez AB, Karas RH, Alsheikh-Ali AA et al. Statins and interstitial lung disease: a systematic review of the literature and of food and drug administration adverse event reports. Chest 2008;134:824–830. doi: 10.1378/chest.08-0943
[92] Etienne D & Reda Y. Statins and their role in acute pancreatitis: Case report and literature review. World J Gastrointest Pharmacol Ther 2014;5:191–195. PMCID: PMC4133445
[93] Singh S & Loke YK. Statins and pancreatitis: a systematic review of observational studies and spontaneous case reports. Drug Saf 2006;29:1123–1132. PMID: 17147459
[94] Preiss D, Tikkanen MJ, Welsh P et al. Lipid-modifying therapies and risk of pancreatitis: a meta-analysis. JAMA 2012;308:804–811. doi: 10.1001/jama.2012.8439
[95] Noel B. Statins and lupus erythematosus. Rheumatology (Oxford) 2004;43:397–398; author reply 398–399. doi: 10.1093/rheumatology/keh035
[96] Knapp AC, Huang J, Starling G et al. Inhibitors of HMG-CoA reductase sensitize human smooth muscle cells to Fas-ligand and cytokine-induced cell death. Atherosclerosis 2000;152:217–227. doi: 10.1016/S0021-9150(99)00462-1