Key points
- Introduction of the ‘precision medicine’ (PM) approach has seen its most significant advances in the field of oncology.
- The key scientific principle behind this approach is to identify predictive biomarkers that can be used to select the most suitable therapeutic agents.
- Biomarkers can be used for diagnostic, prognostic, toxicological and treatment-monitoring purposes.
- Many pharmacogenomic assays have been developed to identify biomarkers.
Cancer learning ‘hub’
Pharmacists are playing an increasingly important role in supporting patients with cancer, working within multidisciplinary teams and improving outcomes. However, in a rapidly evolving field with numbers of new cancer medicines is increasing and the potential for adverse effects, it is now more important than ever for pharmacists to have a solid understanding of the principles of cancer biology, its diagnosis and approaches to treatment and prevention. This new collection of cancer content, brought to you in partnership with BeOne Medicines, provides access to educational resources that support professional development for improved patientIntroduction
Everyone has their own unique genome, which includes small, single nucleotide polymorphisms (SNPs) or large changes in DNA base-pair sequence (e.g. translocation mutations). These can be inherited or introduced during a person’s lifetime owing to the effect of external agents, such as carcinogenic chemicals or radiation. Although usually harmless, these changes can affect the way an individual responds to a therapeutic agent either through differences in the drug target, or through changes to ADME parameters (i.e. absorption, distribution, metabolism and excretion). Precision medicine, sometimes known as ‘personalised medicine’ and abbreviated to ‘PM’[1],[2] , is a term that is increasingly being used to describe treatments tailored to individual patients or groups of patients[3] . The overall goal of the PM approach is to match therapies to individual patients, thus ensuring they receive effective treatment with minimal toxicity. This is particularly important for patients with cancer who may have a limited life expectancy. Furthermore, there has been an ongoing decrease in the cost of sequencing the human genome, which has led to the widespread adoption of integrative sequencing strategies for the study and treatment of cancer[4] . Within the field of oncology, most aspects of a PM approach involve the identification of ‘biomarkers’ associated with a particular cancer type. A biomarker is a mutated nucleic acid sequence, protein or group of proteins, expressed uniquely by the tumour cells (e.g. mutated BRCA1 and BRCA2), or a non-mutated protein or receptor (e.g. HER2 or VEGF) upregulated in tumour cells compared with healthy cells[1] . This review is presented in sections relating to cancer type, although some biomarkers are relevant to more than one. Part 1, which focuses on solid tumours, includes an introduction to the technologies used for companion diagnostic (CDx) tests, along with discussions of the limitations of biomarker testing and the role of regulatory bodies in validating biomarkers and CDx tests. PM-based clinical trials, tumour agnostic anticancer agents, supportive therapies, and funding challenges for the NHS in relation to the growing introduction of novel PM agents and biomarker assays are also covered. Part 2 focuses on haematological cancers, and includes a summary of potential future applications of the PM approach in oncology.Biomarkers, companion diagnostic tests and precision anticancer agents for haematological malignancies
Haematological malignancies are a group of blood-forming cancers that develop in either the bone marrow or the cells of the immune system. Therefore, they are sometimes referred to as blood or liquid tumours[5] . The World Health Organization (WHO) has developed a unified classification system for haematological malignancies, dividing them primarily into neoplastic diseases of the haematopoietic or lymphoid tissues, which are then further divided into sub-categories according to the cells from which they originate[6] . Cancer-relevant biomarkers, the assays to detect and evaluate them, and the associated anticancer drugs have been reviewed based on the primary and patent literature, and information available from conferences and the websites of pharmaceutical companies. The information has been grouped below in sections relating to the type of malignancy. However, although some biomarkers are highly specific for a particular cancer type (e.g. BCR-ABL for chronic myeloid leukaemia [CML])[7] , others are relevant to more than one different type of cancer (e.g. PD-L1 for both myeloma and lymphomas).Leukaemias: chronic lymphocytic, chronic myeloid, acute lymphoblastic and acute myeloid leukaemias
The signal transduction pathways of leukaemias such as CML are activated by the BCR-ABL translocation gene mutation[7] , and patients with this mutation can be selected for treatment with kinase inhibitors such as imatinib (Glivec; Novartis), ponatinib (Iclusig; Incyte Biosciences UK Ltd), bosutinib (Bosulif; Pfizer), dasatinib (Sprycel; Bristol-Meyers Squibb) and nilotinib (Tasigna; Novartis). When patients become resistant to the first agent used for treatment (i.e. typically imatinib) owing to mutations of the amino acid residues within the ATP-binding pocket where these agents interact, treatment can be swapped to one of the second-generation agents that fit with a different orientation in the ATP-binding pocket, thus restoring therapeutic activity (see Figure 1)[8] . 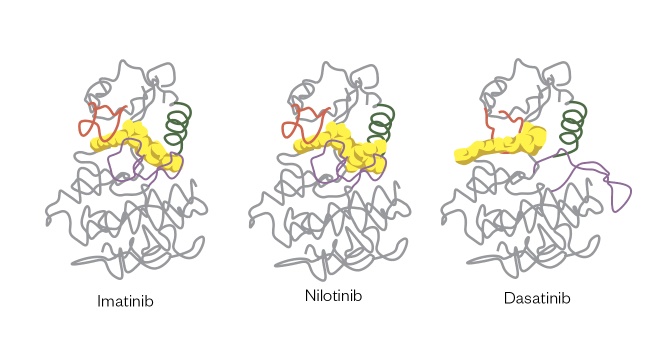
Figure 1: Structures of imatinib, nilotinib and dasatinib interacting in the ATP-binding pocket of the BCR-ABL protein
These interactions result in small but significant changes in binding mode, meaning that nilotinib and dasatinib can be used to treat patients who have become resistant to imatinib owing to small changes in structure within the ATP-binding pocket.
Reproduced with permission from Nat Rev Cancer [8]
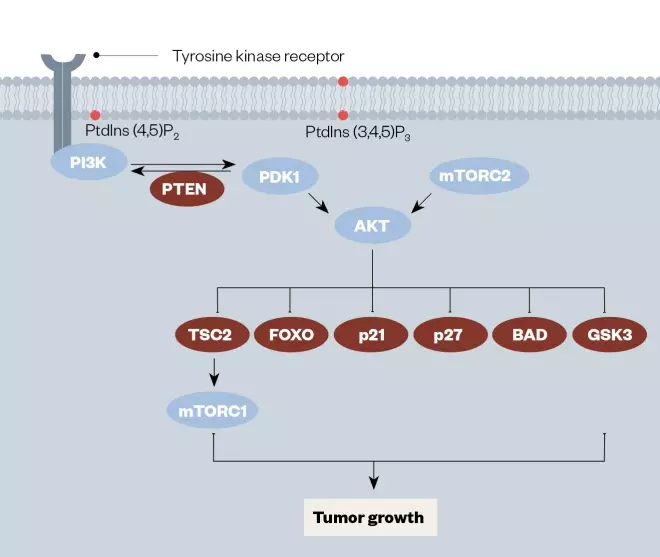
Figure 2: Diagram of the PI3K/PTEN/AkT/mTOR signaling pathway important in leukaemia, with key drug targets highlighted
Reproduced with permission from Mol Cancer Ther [31]
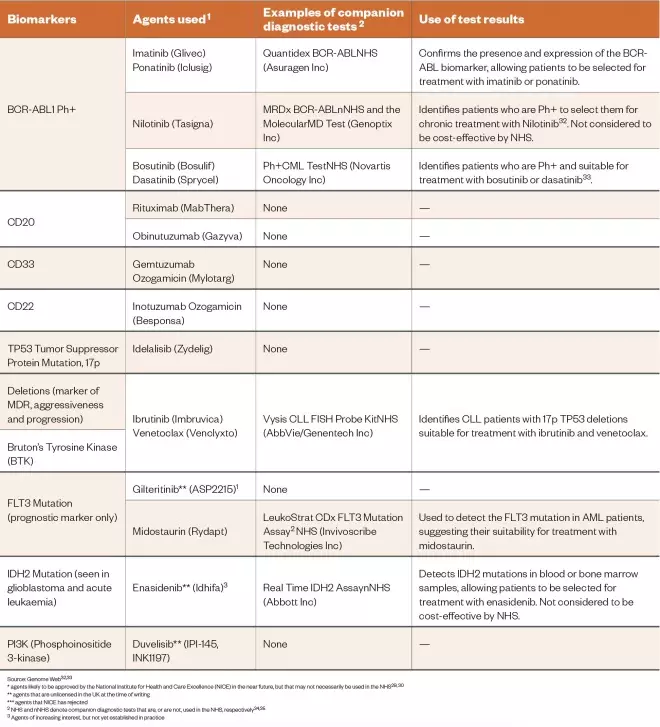
Table 1: Biomarkers associated with the leukaemias the biomarker tests used to identify them and the use of this information for therapy
* agents likely to be approved by the National Institute for Health and Care Excellence (NICE) in the near future, but that may not necessarily be used in the NHS[29],[30]
** agents that are unlicensed in the UK at the time of writing
*** agents that NICE has rejected2 NHS and nNHS denote companion diagnostic tests that are, or are not, used in the NHS, respectively[34],[35]
2 NHS and nNHS denote companion diagnostic tests that are, or are not, used in the NHS, respectively[34],[35]
3 Agents of increasing interest, but not yet established in practice
Lymphomas: Hodgkin and non-Hodgkin lymphoma
Lymphomas are a group of haematological malignancies that derive from lymphocytes and occur predominantly in lymph nodes and other lymphoid organs[36] . There are two main types of lymphomas: Hodgkin and non-Hodgkin, each with their own cancer subtypes. Classical Hodgkin lymphomas are characterised by the presence of Reed–Sternberg cells (mature malignant B cells), whereas non-Hodgkin lymphomas are derived from B- and T-cells, but can arise in the lymph nodes or other surrounding organs[37] . CD20 is used as a biomarker and drug target in B-cell non-Hodgkin lymphoma for targeted radiotherapy and immunotherapy, respectively, for previously untreated relapsed or refractory disease, or in the treatment of relapsed CLL and follicular lymphoma[38] . The agents used to target this biomarker include ibritumomab tiuxetan (Zevalin; Acrotech Biopharma), rituximab (Rituxan; Genentech) and obinutuzumab (Gazyva; Roche)[38] . CD30 is a membrane protein of the tumour necrosis factor receptor and is associated with the Reed–Sternberg cells of classical Hodgkin lymphoma. It plays a role in normal lymphoid interactions, as suggested by its histological detection on the surface of lymphoid cells in reactive lymph nodes, and by its induced expression by purified T- and B-cells following lectin activation[39] . Together with PD-L1, CD30 is one of the biomarkers used in Hodgkin lymphoma, and can be used to select patients for treatment with brentuximab vedotin (Adcetris; Seattle Genetics/Millennium Pharmaceuticals/Takeda). The tumour suppressor protein 53 (TP53) mutation, despite its low frequency in haematological cancers, is considered to be a prognostic biomarker in patients with diffuse large B-cell (DLBCL), follicular and mantle cell lymphomas[36] . For example, TP53 mutation status is associated with poor prognosis in patients with DLBCL[36] . However, Nair et al. have reported that idelalisib (Zydelig, Gilead Sciences) promotes caspase-dependent apoptosis independent of CLL prognostic factors such as mutated TP53[40] . Both TP53 and CD30 are used as part of a second-line diagnosis in Hodgkin lymphoma and cutaneous T-cell lymphoma, as well as in peripheral T-Cell lymphoma (PTCL) patients non-responsive to the CHOP chemotherapy regimen (Cyclophosphamide, Hydroxydaunorubicin [doxorubicin], Oncovin [vincristine] and Prednisolone) used in the treatment of non-Hodgkin lymphoma. Bruton’s tyrosine kinase (BTK) is a biomarker associated with B-cell maturation and is considered to be the major oncogenic driver in some B-cell lymphoid tumour malignancies[41] including non-Hodgkin lymphoma and diffuse large cell B-lymphoma (DLBCL)[41] . The kinase inhibitor ibrutinib (Imbruvica; Janssen-Cilag) works by inhibiting BTK, and has demonstrated efficacy in the activated B-cell-like subtype of diffuse large-cell B-lymphoma but not in the germinal centre (a specialised microstructure that is formed within the follicles of secondary lymphoid tissues such as the spleen and lymph nodes) of B-cell-like subtypes[41] . The PD-1 biomarker is associated with an inferior survival rate for Hodgkin lymphoma regardless of disease status[36] , and its presence is characteristic of nodular lymphocyte-predominant Hodgkin lymphoma. The T-follicular helper lymphocyte cells that rosette around the PD-L1-expressing malignant cells express PD-1, and it is this PD-1/PD-L1 expression that has proven to be of both prognostic and disease-monitoring value[36] . Therefore, anti-PD-1 agents could be useful to treat patients with this type of lymphoma. PI3K is also important in DLBCL[42] . The activity of the PI3K/AkT pathway acts as a prognostic marker[42] , with constitutive activation of the pathway playing a prominent role in regulating the growth and survival of DLBCL cells[43] . There is currently interest in exploiting this biomarker for the treatment of lymphomas. The biomarkers used in both types of lymphomas, the tests used to identify them, and the use of this information in therapy are summarised in Table 2. 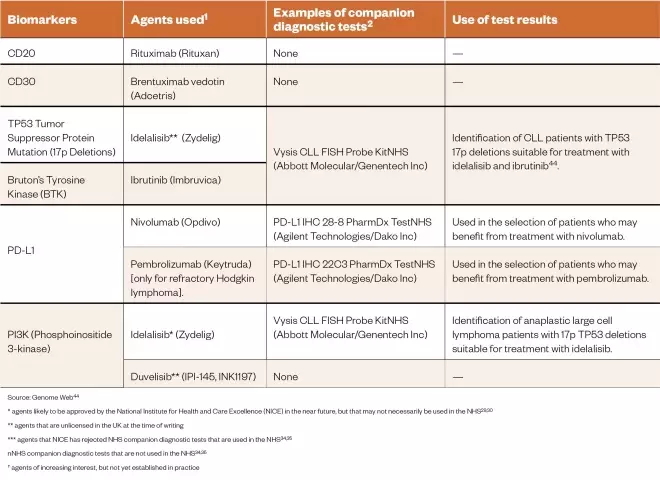
Table 2: The biomarkers used in both types of lymphoma the tests used to identify them and the use of this information in therapy
Source: Genome Web[44]
* agents likely to be approved by the National Institute for Health and Care Excellence (NICE) in the near future, but that may not necessarily be used in the NHS[29],[30]
** agents that are unlicensed in the UK at the time of writing
*** agents that NICE has rejected NHS companion diagnostic tests that are used in the NHS[34],[35]
nNHS companion diagnostic tests that are not used in the NHS[34],[35]
†agents of increasing interest, but not yet established in practice
py
Myeloproliferative neoplasms
These are a group of rare disorders of the bone marrow (including CML) that cause an increase in the number of blood cells[45] . Interest in the Janus family of kinase proteins (JAKs) as potential biomarkers in myeloproliferative neoplasms (MPNs)[46] has shifted from their placement at the centre of multiple point mutation activations in various tumour types, to their production in response to an increase in inflammatory cytokines in the tumour microenvironment produced by infiltrating innate immune cells[47] , to the point where they are now viewed as a biomarker for myeloproliferative diseases. Oncogenic JAKs 1, 2 and 3 are all associated with both lymphoid and myeloid neoplasms[48] . Of particular importance is JAK2V617F, which is prevalent in BCR-ABL1-negative MPN[48] . This mutation affects the non-catalytic pseudo-kinase domain of the Janus family JAK2, thereby diverting its kinase activity[48] . Inhibition of the JAK/STAT3 pathway offers a significant therapeutic benefit due to the capacity of JAK/STAT3 signalling to promote cancer ‘hallmarks’, such as proliferation, survival, angiogenesis and tumour metabolism while suppressing antitumour immunity[47] . Steensma et al. have described the clinical use of JAK2 V616F mutation diagnostic tests in myeloid disorders[49] . They noted that this missense mutation plays an important role in normal haematopoietic growth factor signalling, and is present in patients who are BCR-ABL-negative but have chronic MPN disorder[49] . This mutation causes constitutive activation of the kinase, leading to deregulation of signalling during haematopoietic growth factor stimulation[49] . There are a variety of tools that can detect this mutation, from direct DNA sequencing using allele-specific PCR in either real-time mode or through an amplification refractory mutation system (ARMS), to pyrosequencing[49] . Ruxolitinib (Jakavi, Novartis) was developed to target both JAK1 and JAK2, but is only used for symptom control, reducing both splenomegaly and anaemia. Akpinar et al. sequenced the MPL gene coding the thrombopoietin receptor, and uncovered a new molecular abnormality in JAK2 mutation-negative MPN patients[50] . The most common of these were W515L (tryptophan to leucine) and W515K (tryptophan to lysine) substitutions[50] . These account for only 10% of mutations found in MPN but are present in patients with essential thrombocythemia (ET) and primary idiopathic myelofibrosis (PMF)[50] . Often, patients with W515K and W515L mutations display specific signs and symptoms, including low haemoglobin levels and higher platelet counts at diagnosis as compared with patients with a JAK2 V617F mutation[50] . Another biomarker that has been studied since the 2014 revision of the WHO diagnostic criteria for BCR-ABL1-negative MPN patients is the calreticulin (CALR) mutation[51] . The CALR gene is located on the short arm of chromosome 19, and can contain two different types of mutations, either a 52-bp deletion mutation or a 5-bp TTGTC insertion[51] . Currently, the role of CALR mutations in the molecular pathogenesis of MPN is not entirely clear, but they seem to affect the disease phenotypes of patients with JAK2 mutations and those presenting with ET[51] , [52] . The biomarkers used in MPNs, the tests used to identify them, and the use of this information in therapy are summarised in Table 3. 
Table 3: Biomarkers used in myeloproliferative neoplasms the tests used to identify them and the use of this information in therapy
* agents likely to be approved by the National Institute for Health and Care Excellence (NICE) in the near future, but that may not necessarily be used in the NHS[29],[30] ** agents that are unlicensed in the UK at the time of writing *** agents that NICE has rejected NHS companion diagnostic tests that are used in the NHS[34],[35] nNHS companion diagnostic tests that are not used in the NHS[34],[35] †agents of increasing interest, but not yet established in practice
Multiple myeloma
This is a malignant disorder originating in bone marrow plasma cells, and accounting for 13% of all haematological cancers[53] . In multiple myeloma (MM), malignant plasma cells undergo a massive clonal expansion resulting in the production of a high level of monoclonal immunoglobulin[54] . These cells proliferate rapidly and infiltrate the bone marrow, interfering with cell signalling pathways involved in osteoblast formation. Novel therapies used to treat this malignancy have been reviewed, together with the role of biomarkers as tools for imaging and diagnosis[55] . Treatment starts with the identification of symptoms and co-morbidities, leading to either an autograft or the start of one to three lines of therapeutics. Cereblon (CRBN) is a protein encoded by the CRBN gene, and is a target for immunomodulatory drugs (IMDs) such as lenalidomide (Revlimid; Celgene Ltd) and pomalidomide (Imnovid; Celgene Ltd)[56] ,[57] , key agents within the various treatment strategies for multiple haematologic malignancies including MM. It has been suggested that over-expression of cereblon could serve as a predictive biomarker for resistance to IMDs, which could be useful to establish prior to initiating therapy, although this remains controversial[56] . The CRBN -associated transcription factors ikaros and aiolos are considered by some researchers to have more potential as functional biomarkers since they appear as downstream substrates in the transcription of this protein[56] . HR23B is another potential biomarker for MM that has an important role in transporting ubiquitinated cargo proteins to the proteasome[58] . Khan et al. have reported that HR23B governs the sensitivity of tumour cells to histone deacetylase inhibitors such as vorinostat (Zolinza; MSD) and panobinostat (Farydak; Novartis)[58] . Thus, it can be used as a predictive biomarker in this clinical setting. Using novel technologies such as 2D-DIGE and mass spectroscopy (MS)[59] , Rajpal et al. have described biomarkers found only in MM patients responding to thalidomide (Thalidomide; Celgene). These biomarkers, which include ZAG, VDB, SAA, B2M and Hp, are found at levels statistically different compared with non-responders[59] . They comprise a combination of serum protein biomarkers (e.g. ZAG, VDB and SAA) present on the surface of many cells including lymphocytes, a highly prognostic factor in combination with albumin, and an apolipoprotein synthesised in response to inflammatory biomarkers secreted by activated monocytes and macrophages[59],[60] . The interactions between chemokines such as stromal cell-derived factor-1 and its receptor (chemokine C-X-X motif receptor-4) play a central role in MM including effects on cellular proliferation, phosphorylation of mitogen-activated protein kinase1/2 (MAPK) and p42/44 MAPK, induction of interleukin-6 and VEGF secretion[61] . Therefore, this interaction can be used as a potential biomarker for diagnosing MM. It has also been used to develop proteasome inhibitors such as bortezomib (Velcade; Janssen-Cilag Ltd) and carfilzomib (Kyprolis; Amgen Ltd) which work by inhibiting the interaction between stromal cell-derived factor-1 and its receptor C-X-X motif[62] . The sensitivity to these inhibitors is increased through the mobilisation of MM cells from the bone marrow microenvironment[63] . The reduction in overall expression of CXCR4 in bortezomib-resistant cells has led to the conclusion that CXCR4 could be used as a potential diagnostic biomarker for predicting patient survival in relation to treatment with these drugs[62],[63] . CD38 is a 46kDa transmembrane glycoprotein with a role in adhesion, signalling events and bifunctional ectoenzymatic activities contributing to the mobilisation of intracellular calcium[64] . It is present in low levels in myeloid cells under normal circumstances but is over-expressed when the cells become malignant[64] . Therefore, CD38 has potential use as both a diagnostic biomarker and as a therapeutic target for this disease[64] . Daratumumab (Darzalex; Janssen-Cilag) is a human IgG1k monoclonal antibody that binds to CD38[65] , and pre-clinical studies have shown that it induces target-cell killing through a number of mechanisms including complement-mediated and antibody-dependent cell-mediated cytotoxicity (ADCC) events, antibody-dependent cellular phagocytosis, apoptosis and inhibition of the enzymatic activity of CD38[65] , all of which occur at very low concentrations and induce a potent cytotoxic response[64] . Elotuzumab (Empliciti; Bristol-Meyers Squibb) is a humanised IgG1k monoclonal antibody that has been approved by the FDA for the treatment of MM[66] . It targets the signalling lymphocyte activation molecule family member 7 (SLAMF7), and works by enhancing the cytotoxic activity of natural killer cells (NKCs) (see Figure 3)[67] . SLAMF7 is expressed by a number of cell types including MM cells, NKCs, leukocytes and plasma cells[66],[67] . Studies by Wang et al. have shown that SLAMF7-mediated signalling is important for the adhesive interaction between myeloma cells and bone marrow stromal cells (BMSC), a process that activates the ERK1/2, STAT3 and AKT signalling pathways[66] . Therefore, SLAMF7 is considered to be an ideal therapeutic target for MM[66] . As a complication of their disease, many MM patients experience skeletal problems such as bone pain, hypercalcaemia and pathological fractures resulting from secondary or lytic bone lesions[68] . Studies by Roux et al. have suggested that these problems may be exacerbated by excessive production of osteoclast-activating factors produced either by the bone marrow itself or by the surrounding microenvironment[68],[69] . The main factor expressed by developing osteoclasts is receptor activator of nuclear factor kappa-Î’ ligand (RANKL)[68],[69] . Since MM is characterised by a high rate of bone resorption, it is useful for clinicians to evaluate and monitor RANKL expression. The human monoclonal antibody denosumab (Xgeva; Amgen) is targeted to RANKL, and so is used to treat patients with bone disease[70] . The biomarkers used in myeloma, the tests used to identify them, and the use of this information in therapy are summarised in Table 4. 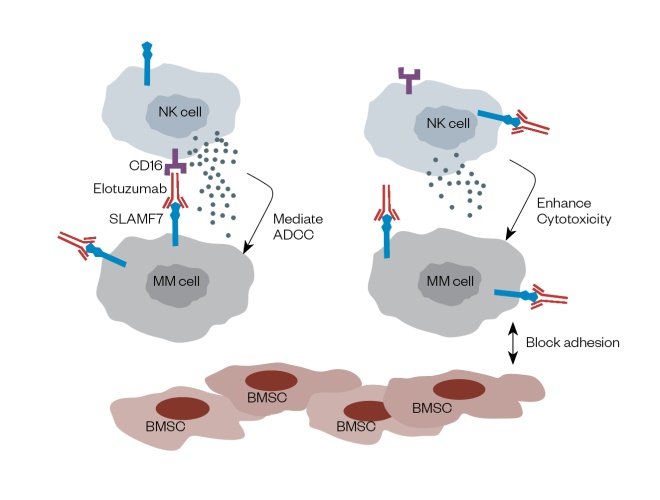
Figure 3: Diagram of the mechanism of action of Elotuzuma b (Empliciti)
Reproduced with permission from J Haematol Oncol [66]

Table 4: The biomarkers used in myeloma the tests used to identify them and the use of this information for therapy
Source: Sebia, Janssen Biotech Ink multiple myeloma IVD test agreement[71]
* agents likely to be approved by the National Institute for Health and Care Excellence (NICE) in the near future, but that may not necessarily be used in the NHS[29],[30]
** agents that are unlicensed in the UK at the time of writing
*** agents that NICE has rejected NHS companion diagnostic tests that are used in the NHS[34],[35]
nNHS companion diagnostic tests that are not used in the NHS[34],[35]
†agents of increasing interest, but not yet established in practice
Supportive therapies
Supportive therapies are used to offset the side effects frequently experienced with most types of anticancer therapies, including PMs, and this was discussed in detail in relation to solid tumours in part 1 of this review. Although most of this previous discussion is relevant, with haematological cancers immunosuppression can result from the immune defects of the underlying disease. In addition, the chemotherapy regimens used for haematological cancers are often very intensive, further increasing the risk of infection[72] . Therefore, supportive therapies for these patients can include platelets, blood infusions, immunoglobulins and drug therapies including antibiotics [72] . As with solid tumour patients, anti-emetics are often required pre- and post-treatment to reduce nausea and vomiting[73] . Other medicines used specifically for haematology patients include allopurinol to prevent tumour-lysis syndrome, co-trimoxazole to prevent infection with pneumocystis pneumonia, and aspirin or a low molecular weight heparin to reduce the risk of thrombosis[73] .Future strategies in the precision medicine approach
There are several visions for the PM approach in the longer term. First, the development of accurate and selective non-invasive tumour biomarker screens, and their widespread and regular use by healthy individuals (e.g. the identification of biomarkers in urine or finger-prick blood samples), should lead to the very early diagnosis of most cancers along which effective treatments (hopefully ‘cures’ in many cases). Another vision is that, for cancers that do develop, new generations of PM-based agents might be used to treat most cancer types as chronic diseases, thus allowing effective treatment and long-term survival through simple oral dosing, a precedent set with agents such as imatinib. Ultimately, whole genome sequencing (WGS) could be carried out at birth (potentially even in utero) for the entire population, and genetic biomarkers used to predict cancer risk for everyone, with prophylactic intervention if appropriate. However, the ethical implications of this approach are not insignificant and may be considered more of a barrier than the practicalities. Although space does not permit a detailed discussion of recent advances in the development of techniques to identify and quantify biomarkers in blood or urine, some examples of the development of methods to detect and measure tumour miRNAs and DNA in blood samples are briefly described below. Methods are also being developed to isolate tumour cells from blood and urine samples, which should not only allow identification of the cancer type, but may also produce biomarker information from their DNA and cellular contents, thus allowing early PM drug selection and treatment. A significant research effort is underway to develop methodologies to detect cancer-relevant biomarkers in small samples of blood or urine so that various cancer types might be diagnosed early through a simple, minimally invasive test. For example, microRNAs (miRNAs) are being investigated as a new class of small non-coding RNAs that regulate gene expression at a post-transcriptional level by either blocking or degrading the translation of messenger RNA targets[74] . MiRNAs have been identified in the blood of cancer patients, and this discovery has led to the use of miRNAs as important cancer biomarkers. MiRNAs appear to have multifunctional roles, and can be used to help distinguish between healthy and malignant cells[74] , in identifying the origin of secondary (i.e. metastatic) tumours[74] and in distinguishing between different subtypes of cells from the same tumour[74] . Two recent studies have focused on the detection of circulating miRNAs in blood as specific methods for cancer detection[53],[75] . These dysregulated miRNAs were found in the blood of patients who had monoclonal gammopathy of unetermined significance (MGUS) and myeloma[53],[74] . In addition, researchers in Japan have recently developed a new approach to non-invasive liquid biopsies using nanowires to extract thousands of urinary miRNAs from just 1 milliliter of urine[76] . This approach was used to successfully discriminate between samples from patients with pancreatic, prostate and bladder cancer and those collected from healthy individuals[76] , although further work is required to fully validate this approach. Another rapidly advancing technology involves the detection of circulating DNA in blood. For example, a study in 2017 demonstrated the possibility of using circulating tumour DNA to detect MM. In this approach the use of liquid-biopsy sequencing (LB-Seq) helped to uncover a large set of MM mutations[77],[78] , and it was established that 96% of the mutations could be accurately detected by the genetic profiling of matched bone-marrow-derived tumour DNA with more than 98% specificity[78] . These results suggest that LB-Seq might be used in the future as a complementary method to invasive bone marrow aspiration-based testing, perhaps eventually replacing it. More recently, technologies have been developed to detect a combination of circulating proteins and mutated cell-free DNA in blood[79] as a means to detect cancer as early as possible through a simple finger-prick test[80] . For example, researchers at Johns Hopkins have developed a test, known as CancerSEEK[80] capable of analysing circulating tumour DNA and proteins together for the detection of eight common surgically resectable, non-metastatic cancer types including ovary, liver, stomach, pancreas, oesophagus, colorectal, lung and breast[81],[82] . In a study involving 1,005 patients, the results were positive in a median of 70% of the eight cancer types[82] . The sensitivities ranged from 69% to 98% for the detection of five cancer types (i.e. ovary, liver, stomach, pancreas and esophagus) for which there are presently no screening tests available for average-risk individuals. The specificity of CancerSEEK was >99%, and only 7 out of 812 control samples from healthy individuals generated a false positive result. In addition, CancerSEEK localised the cancer to a small number of anatomic sites in a median of 83% of the patients. Other areas of research are focusing on the development of advanced (e.g. multiplexed) assays to analyse multiple genes in biopsied tumour tissue for purposes including therapy selection and prognosis. For example, the FoundationOne CDx (F1CDx) test utilises next-generation sequencing (NGS) to detect variants in up to 324 genes, also identifying two key signatures, microsatellite instability[65] and tumour mutational burden (TMB) across solid tumours of many different types[83] . In addition to its use in NSCLC, melanoma, breast, colorectal and ovarian cancers, it has been used as a companion diagnostic test for 17 FDA-approved targeted treatments[83] including the anti-PD 1 immuno-oncology agent pembrolizumab (Keytruda; MSD)[84] , which is discussed in more detail in part 1 of this review.Conclusion
In the past three decades the PM approach to cancer therapy has progressed from the concept phase to practical utility. This has significant implications for healthcare systems such as the NHS, in that it creates the opportunity to provide anticancer therapies for patients with a higher degree of certainty of providing clinical benefit[79] . However, these new therapies are expensive and require CDx tests to select patients, which has cost implications. Although pharmaceutical companies are adjusting to this new paradigm by setting higher prices for novel PM-based anticancer agents that will sell in lower volume, government-funded healthcare organisations such as the NHS will come under increasing pressure to fund these new therapies because of their clinical effectiveness. For these new PM approaches, the growing burden on clinicians, pharmacists and other healthcare professionals will become increasingly significant owing to the additional requirements for the selection of patients using CDx tests, followed by appropriate drug selection and dose scheduling based on the test results[80] . The complexity of this new treatment paradigm demands that healthcare professionals not only have the technical expertise and knowledge to deliver it, but also have the necessary communication skills to explain diagnostic test results and drug selection decisions clearly and confidently to patients and other healthcare professionals. This review provides concise information on the current status of the PM approach in the diagnosis and therapy of haematological malignancies, and should be of use to clinicians, pharmacists and other healthcare professionals. Although this part 2 of the review has focused on haematological cancers, part 1 reviewed the PM approach to the treatment of solid tumours. Finally, it should be noted that the accuracy and coverage of this review (and Part 1 which covered solid tumours) will be short-lived owing to the rapid progress being made in this area, with new agents and CDx tests entering the clinic at a remarkable rate. By necessity, each paragraph of this review represents only a brief summary of what ideally should be a systematic review of thousands of publications. The reader should be aware of this, and is encouraged to look further into the literature in their area of interest.References
[1] Biddle N & Smith ML. Biomarkers in cancer: an introductory guide for advocates: Research Advocacy Network; 2010. Available at: http://www.researchadvocacy.org/sites/default/files/resources/BiomarkerinCancer_WebDownloadVersion.pdf (accessed February 2020)
[2] Brain cancer coalition plans precision medicine study. GenomeWeb. 2017. Available at:https://www.genomeweb.com/cancer/brain-cancer-coalition-plans-precision-medicine-study?utm_source=SilverpopMailing&utm_medium=email&utm_campaign=Cancer%20Bulletin:%20One%20Year%20In,%20GenomeDx%20Confident%20in%20Market%20Share%20for%20Prostate%20Biopsy%20Dx%20-%2002/06/2017%2004:15:00%20PM#.XjQE7C-cbjA (accessed February 2020)
[3] Sciences AoM. Stratified, personalised or P4 medicine: a new direction for placing the patient at the centre of healthcare and health education. University of Southampton; 2015.
[4] Robinson DR, Wu YM, Lonigro RJ et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548(7667):297–303. doi: 10.1038/nature23306
[5] Hematologic cancer. National Cancer Institute. Available at: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/hematologic-cancer (accessed February 2020)
[6] Jurlander J. Hematological malignancies, leukemias and lymphomas. In: Schwab M, editor. Encyclopedia of cancer. Springer Berlin Heidelberg; Berlin. 2011;1640–1644.
[7] Salesse S & Verfaillie CM. BCR/ABL: from molecular mechanisms of leukemia induction to treatment of chronic myelogenous leukemia. Oncogene 2002;21(56):8547–8559. doi: 10.1038/sj.onc.1206082
[8] Weisberg E, Manley PW, Cowan-Jacob SW et al. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer 2007;7(5):345–356. doi: 10.1038/nrc2126
[9] Tamayose K, Sato N, Ando J et al. CD3-negative, CD20-positive T-cell prolymphocytic leukemia: case report and review of the literature. Am J Hematol 2002;71(4):331–335. doi: 10.1002/ajh.10224
[10] Ofatumumab. Specialist Pharmacy Service. 2019. Available at: https://www.sps.nhs.uk/medicines/ofatumumab (accessed February 2020)
[11] PharmaLetter. BRIEF — Amid low sales, Arzerra for CLL withdrawn from ex-US markets. 2018. Available at: https://www.thepharmaletter.com/in-brief/brief-amid-low-sales-arzerra-for-cll-withdrawn-from-ex-us-markets (accessed February 2020)
[12] Walter RB. The role of CD33 as therapeutic target in acute myeloid leukemia. Expert Opin Ther Tar 2014;18(7):715–718. doi: 10.1517/14728222.2014.909413
[13] De Propris MS, Raponi S, Diverio D et al. High CD33 expression levels in acute myeloid leukemia cells carrying the nucleophosmin (NPM1) mutation. Haematologica 2011;96(10):1548–1551. doi: 10.3324/haematol.2011.043786
[14] Walter RB, Appelbaum FR, Estey EH & Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012;119(26):6198–6208. doi: 10.1182/blood-2011-11-325050
[15] Kantarjian H, Thomas D, Jorgensen J et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer 2013;119(15):2728–2736. doi: 10.1002/cncr.28136
[16] Dijoseph JF, Dougher MM, Armellino DC, Evans DY & Damle NK. Therapeutic potential of CD22-specific antibody-targeted chemotherapy using inotuzumab ozogamicin (CMC-544) for the treatment of acute lymphoblastic leukemia. Leukemia 2007;21(11):2240–2245. doi: 10.1038/sj.leu.2404866
[17] Chiaretti S, Brugnoletti F, Tavolaro S et al. TP53 mutations are frequent in adult acute lymphoblastic leukemia cases negative for recurrent fusion genes and correlate with poor response to induction therapy. Haematologica 2013;98(5):e59–61. doi: 10.3324/haematol.2012.076786
[18] Ray T. AbbVie Venclexta, Abbott CDx approval provide docs opportunity to individualize CLL treatment. GenomeWeb. 2016. Available at: https://www.genomeweb.com/molecular-diagnostics/abbvie-venclexta-abbott-cdx-approval-provide-docs-opportunity-individualize (accessed February 2020)
[19] Woyach JA, Bojnik E, Ruppert AS et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014;123(8):1207–1213. doi: 10.1182/blood-2013-07-515361
[20] Quentmeier H, Reinhardt J, Zaborski M & Drexler HG. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia 2003;17(1):120–124. doi: 10.1038/sj.leu.2402740
[21] Gilliland DG & Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002;100(5):1532–1542. doi: 10.1182/blood-2002-02-0492
[22] Gilliland DG & Griffin JD. Role of FLT3 in leukemia. Curr Opin Hematol 2002;9(4):274-281. doi: 10.1097/00062752-200207000-00003
[23] National Institute for Health and Care Excellence. Midostaurin for untreated acute myeloid leukaemia. Technology appraisal guidance [TA523]. 2018. Available at: https://www.nice.org.uk/guidance/ta523 (accessed February 2020)
[24] Mondesir J, Willekens C, Touat M & de Botton S. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J Blood Medicine 2016;7:171–180. doi: 10.2147/JBM.S70716
[25] Parker SJ & Metallo CM. Metabolic consequences of oncogenic IDH mutations. Pharmacol Ther 2015;152:54–62. doi: 10.1016/j.pharmthera.2015.05.003
[26] Abbott receives FDA approval for IDH2 mutation CDx alongside AML drug. GenomeWeb. 2017. Available at: https://www.genomeweb.com/molecular-diagnostics/abbott-receives-fda-approval-idh2-mutation-cdx-alongside-aml-drug?utm_source=Sailthru&utm_medium=email&utm_campaign=GWDN%20Tues%20PM%202017-08-01&utm_term=GW%20Daily%20News%20Bulletin (accessed February 2020)
[27] Steelman LS, Abrams SL, Whelan J et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008;22(4):686–707. doi: 10.1038/leu.2008.26
[28] Park S, Chapuis N, Tamburini J et al. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010;95(5):819–828. doi: 10.3324/haematol.2009.013797
[29] Yang Q, Modi P, Newcomb T, Queva C & Gandhi V. Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res 2015;21(7):1537–1542. doi: 10.1158/1078-0432.CCR-14-2034
[30] FDA accepts new drug application for Duvelisib and grants priority review. BioSpace. 2018. Available at: https://www.biospace.com/article/releases/fda-accepts-new-drug-application-for-duvelisib-and-grants-priority-review (accessed February 2020)
[31] Dienstmann R, Rodon J, Serra V & Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther 2014;13(5):1021–1031. doi: 10.1158/1535-7163.MCT-13-0639
[32] MolecularMD gets FDA authorization for companion Dx to Novartis CML drug. GenomeWeb. 2017. Available at: https://www.genomeweb.com/companion-diagnostics/molecularmd-gets-fda-authorization-companion-dx-novartis-cml-drug?utm_source=Sailthru&utm_medium=email&utm_campaign=GWDN%20Wed%20AM%202017-12-27&utm_term=GW%20Daily%20News%20Bulletin#.XjP-ay-cbjA (accessed February 2020)
[33] MolecularMD, Genoptix Ink marketing agreement for CDx to Novartis CML drug. GenomeWeb. 2018. Available at: https://www.genomeweb.com/pcr/molecularmd-genoptix-ink-marketing-agreement-cdx-novartis-cml-drug?utm_source=Sailthru&utm_medium=email&utm_campaign=GWDN%20Mon%20AM%202018-06-04&utm_term=GW%20Daily%20News%20Bulletin#.XjP-OC-cbjA (accessed February 2020)
[34] Viapath. Test index. Viapath. 2014. Available at: http://www.viapath.co.uk/tests-index (accessed February 2020)
[35] Viapath. New tests. Viapath. 2013. Available at: http://www.viapath.co.uk/new-tests (accessed February 2020)
[36] Sun R, Medeiros LJ & Young KH. Diagnostic and predictive biomarkers for lymphoma diagnosis and treatment in the era of precision medicine. Mod Pathol. 2016;29:1118–1142. doi: 10.1038/modpathol.2016.92
[37] Freedman A. What is the difference between Hodgkin lymphoma and non-Hodgkin lymphoma? Dana-Farber Cancer Institute. 2015. Available at: http://blog.dana-farber.org/insight/2015/07/what-is-the-difference-between-hodgkin-lymphoma-and-non-hodgkin-lymphoma (accessed February 2020)
[38] Pillai RK, Nathwani BN & Yang L. Predictive biomarkers and targeted therapies for lymphoid malignancies. In: Predictive Biomarkers in Oncology. 2018. Available at: https://link.springer.com/chapter/10.1007/978-3-319-95228-4_32 (accessed February 2020)
[39] Smith H CA, Farrah T, Baker E et al. CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell 1993;73:1349–1360. doi: 10.1016/0092-8674(93)90361-S
[40] Nair KS & Cheson B. The role of idelalisib in the treatment of relapsed and refractory chronic lymphocytic leukemia. Ther Adv Hematol 2016;7(2):69–84. doi: 10.1177/2040620715625966
[41] Wang JJ, Zheng M, Huang X et al. Abstract 2167: efficacy assessment of BTK inhibitor ibrutinib in de novo and viral-induced B cell lymphoma. Cancer Res 2018;78(13 Supplement):2167. doi: 10.1158/1538-7445.Am2018-2167
[42] Huang X, Shen Y, Liu M et al. Quantitative proteomics reveals that miR-155 regulates the PI3K-AKT pathway in diffuse large B-cell lymphoma. Am J Pathol 2012;181(1):26–33. doi: 10.1016/j.ajpath.2012.03.013
[43] Uddin S, Hussain AR, Siraj AK et al. Role of phosphatidylinositol 3’-kinase/AKT pathway in diffuse large B-cell lymphoma survival.Blood 2006;108(13):4178–4186. doi: 10.1182/blood-2006-04-016907
[44] Ray T. FDA approves Imbruvica for subset of CLL patients with 17p deletions. GenomeWeb. 2014. Available at: https://www.genomeweb.com/clinical-genomics/fda-approves-imbruvica-subset-cll-patients-17p-deletions (accessed February 2020)
[45] Myeloproliferative neoplasms. Cancer Research UK. 2017. Available at: http://www.cancerresearchuk.org/about-cancer/other-conditions/myeloproliferative-neoplasms (accessed February 2020)
[46] Kuchenbaecker KB, Hopper JL, Barnes DR et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017;317(23):2402. doi: 10.1001/jama.2017.7112
[47] Buchert M, Burns CJ & Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 2016;35(8):939–951. doi: 10.1038/onc.2015.150
[48] Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 2010;24(6):1128–1138. doi: 10.1038/leu.2010.69
[49] Steensma DP. JAK2 V617F in myeloid disorders: molecular diagnostic techniques and their clinical utility: a paper from the 2005 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn 2006;8(4):397–411. doi: 10.2353/jmoldx.2006.060007
[50] Akpinar TS, Hancer VS, Nalcaci M & Diz-Kucukkaya R. MPL W515L/K Mutations in Chronic Myeloproliferative Neoplasms. Turk J Haematol 2013;30(1):8–12. doi: 10.4274/tjh.65807
[51] Luo W & Yu Z. Calreticulin (CALR) mutation in myeloproliferative neoplasms (MPNs). Stem Cell Investig 2015;2(16):16. doi: 10.3978/j.issn.2306-9759.2015.08.01
[52] Lim KH, Chang YC, Gon-Shen Chen C et al. Frequent CALR exon 9 alterations in JAK2 V617F-mutated essential thrombocythemia detected by high-resolution melting analysis. Blood Cancer J 2015;5(e295):e295. doi: 10.1038/bcj.2015.21
[53] Jones CI, Zabolotskaya MV, King AJ et al. Identification of circulating microRNAs as diagnostic biomarkers for use in multiple myeloma. Br J Cancer 2012;107(12):1987–1996. doi: 10.1038/bjc.2012.525
[54] Klewes L, Vallente R, Dupas E et al. Three-dimensional nuclear telomere organization in multiple myeloma. Transl Oncol 2013;6(6):749–756. doi: 10.1593/tlo.13613
[55] Piazzi G, Acosta J, Smith B, Faye N et al. Multiple myeloma overview: novel therapies, the role of biomarkers and imaging, operational aspects. Online: QuintilesIMS; 2016. Available at: http://www.mediantechnologies.com/wp-content/uploads/2017/01/Multiple-Myeloma-Overview-WP-Final.pdf (accessed February 2020)
[56] Schuster SR, Kortuem KM, Zhu YX et al. The clinical significance of cereblon expression in multiple myeloma. Leukemia Res 2014;38(1):23–28. doi: 10.1016/j.leukres.2013.08.015
[57] Lopez-Girona A, Mendy D, Ito Tet al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012;26(11):2326–2335. doi: 10.1038/leu.2012.119
[58] Khan O, Fotheringham S, Wood V et al. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci USA 2010;107(14):6532–6537. doi: 10.1073/pnas.0913912107
[59] Rajpal R, Dowling P, Meiller J et al. A novel panel of protein biomarkers for predicting response to thalidomide-based therapy in newly diagnosed multiple myeloma patients. Proteomics 2011;11(8):1391–1402. doi: 10.1002/pmic.201000471
[60] Skates SJ, Greene MH, Buys SS et al. Early detection of ovarian cancer using the risk of ovarian cancer algorithm with frequent CA125 testing in women at increased familial risk — combined results from two screening trials. Clin Cancer Res 2017;23(14). doi: 10.1158/1078-0432.ccr-15-2750
[61] Azab AK, Runnels JM, Pitsillides C et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113(18):4341–4351. doi: 10.1182/blood-2008-10-186668
[62] Fall DJ, Stessman H, Patel SS et al. Utilization of translational bioinformatics to identify novel biomarkers of bortezomib resistance in multiple myeloma. J Cancer 2014;5(9):720–727. doi: 10.7150/jca.9864
[63] Stessman HA, Mansoor A, Zhan F et al. Reduced CXCR4 expression is associated with extramedullary disease in a mouse model of myeloma and predicts poor survival in multiple myeloma patients treated with bortezomib. 2013;27(10):2075–2077. doi: 10.1038/leu.2013.148
[64] de Weers M, Tai YT, van der Veer MS et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol 2011;186(3):1840–1848. doi: 10.4049/jimmunol.1003032
[65] Lokhorst HM, Plesner T, Laubach JP et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015;373(13):1207–1219. doi: 10.1056/NEJMoa1506348
[66] Wang Y, Sanchez L, Siegel DS & Wang ML. Elotuzumab for the treatment of multiple myeloma. J Hematol Oncol 2016;9(55):1–8. doi: 10.1186/s13045-016-0284-z
[67] van de Donk NW, Moreau P, Plesner T et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood 2016;127(6):681–695. doi: 10.1182/blood-2015-10-646810
[68] Roux S, Meignin V, Quillard J et al. RANK (receptor activator of nuclear factor-kappaB and RANKLexpression in multiple myeloma. Br J Haematol 2002;117(1):86–92. doi: 10.1046/j.1365-2141.2002.03417.x
[69] Giuliani N, Colla S, Sala R et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappaB ligand (RANKL) in T lymphocytes: a potential role in multiple myeloma bone disease. Blood 2002;100(13):4615–4621. doi: 10.1182/blood-2002-04-1121
[70] Body JJ, Facon T, Coleman RE et al. A study of the biological receptor activator of nuclear factor-kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res 2006;12(4):1221–1228. doi: 10.1158/1078-0432.CCR-05-1933
[71] Sebia, Janssen Biotech Ink multiple myeloma IVD test agreement. 360Dx; New York. 2017. Available at: https://www.360dx.com/business-news/sebia-janssen-biotech-ink-multiple-myeloma-ivd-test-agreement (accessed February 2020)
[72] Hiemenz J & Munker R. Supportive care in hematology. Totowa, New Jersey: Humana Press; 2007
[73] Gunduz E & Gulbas Z. Supportive care in hemato-oncology: a review in light of the latest guidelines. Turk J Haematol 2012;29(1):1–9. doi: 10.5505/tjh.2012.10327
[74] Wittmann J & Jack HM. Serum microRNAs as powerful cancer biomarkers. Biochim Biophys Acta 2010;1806(2):200–207. doi: 10.1016/j.bbcan.2010.07.002
[75] Kubiczkova L, Kryukov F, Slaby O et al. Circulating serum microRNAs as novel diagnostic and prognostic biomarkers for multiple myeloma and monoclonal gammopathy of undetermined significance. Haematologica 2014;99(3):511–518. doi: 10.3324/haematol.2013.093500
[76] Petrone J. Japanese team develops nanowire technology for detecting cancer biomarkers in urine. GenomeWeb. 2017. Available at: https://www.genomeweb.com/molecular-diagnostics/japanese-team-develops-nanowire-technology-detecting-cancer-biomarkers-urine?utm_source=Sailthru&utm_medium=email&utm_campaign=GW%20Cancer%20Mon%202017-12-18&utm_term=Cancer%20Bulletin#.XjPtsC-cbjA (accessed February 2020)
[77] Multiple myeloma study suggests circulating DNA test complements bone marrow testing. GenomeWeb. 2017. Available at: https://www.genomeweb.com/sequencing/multiple-myeloma-study-suggests-circulating-dna-test-complements-bone-marrow-testing?utm_source=Sailthru&utm_medium=email&utm_campaign=GWDN%20Mon%20PM%202017-06-05&utm_term=GW%20Daily%20News%20Bulletin#.XjPtWi-cbjA (accessed February 2020)
[78] Kis O, Kaedbey R, Chow S et al. Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates. Nat Commun 2017;8(15086):15086. doi: 10.1038/ncomms15086
[79] Gisela CaÌceres P, Puskas JA & Magliocco AM. Circulating tumor cells: a window into tumor development and therapeutic effectiveness. Cancer Control 2015;22(2):167–176. doi: 10.1177/107327481502200207
[80] Heath JR, Davis ME & Hood L. Nanomedicine targets CANCER: viewing each human body as a system of interacting molecular networks and targeting disruptions in the system with nanoscale technologies can transform how disease is understood, attacked and possibly prevented. Sci Am 2009;300(2):44–51. PMID: 19186705
[81] Gilmore J. Johns Hopkins-led team develops blood test to detect multiple early-stage cancers. GenomeWeb. 2018. Available at: https://www.genomeweb.com/immunoassays/johns-hopkins-led-team-develops-blood-test-detect-multiple-early-stage-cancers?utm_source=Sailthru&utm_medium=email&utm_campaign=GWCancer Thurs 2018-01-18&utm_term=Cancer Bulletin (accessed February 2020)
[82] Cohen JD, Li L, Wang Y et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018:359(6378)1–10. doi: 10.1126/science.aar3247
[83] Foundation Medicine gains FDA approval, CMS coverage proposal for NGS cancer profiling test. GenomeWeb. 2017. Available at: https://www.genomeweb.com/molecular-diagnostics/foundation-medicine-gains-fda-approval-cms-coverage-proposal-ngs-cancer (accessed February 2020)
[84] Merck, Foundation Medicine to develop companion diagnostics for immunotherapy. GenomeWeb. 2018. Available at: https://www.genomeweb.com/cancer/merck-foundation-medicine-develop-companion-diagnostics-immunotherapy?utm_source=Sailthru&utm_medium=email&utm_campaign=GWDN Thurs AM 2018-05-24&utm_term=GW Daily News Bulletin (accessed February 2020)