Abstract
In the era of precision medicine, timely cancer genotyping is necessary to guide treatment and glean valuable prognostic information. Invasive tissue sampling is the standard practice to establish a cancer diagnosis but has limitations. Circulating tumour DNA (ctDNA) refers to cell-free DNA that originates from tumour. ctDNA assays are minimally invasive alternatives to tissue-based molecular tests and are already used in the management of many advanced cancer subtypes. Future employments of ctDNA include: cancer screening; minimal residual disease detection; therapy-response monitoring; identification of resistance mechanisms; and cancer clonal evaluation. In this article, we discuss the current and future applications of ctDNA in common solid tumours, treatment implications, and the importance of the molecular tumour board.
Key words: ctDNA; targeted therapy; next generation sequencing; precision medicine
Key points
- Circulating tumour DNA (ctDNA) refers to cell-free DNA that originates from tumour;
- Assessing ctDNA in plasma is minimally invasive, requiring a simple blood draw;
- The most frequent application of ctDNA is the genotyping of advanced malignancies;
- ctDNA results have high concordance rates with tissue results and can be used to guide targeted therapies;
- Molecular tumour boards can be utilised to interpret ctDNA results;
- Future uses of ctDNA include assessing treatment response, minimal residual disease monitoring and screening.
Cancer learning ‘hub’
Pharmacists are playing an increasingly important role in supporting patients with cancer, working within multidisciplinary teams and improving outcomes.
However, in a rapidly evolving field with numbers of new cancer medicines is increasing and the potential for adverse effects, it is now more important than ever for pharmacists to have a solid understanding of the principles of cancer biology, its diagnosis and approaches to treatment and prevention.
This new collection of cancer content, brought to you in partnership with BeOne Medicines, provides access to educational resources that support professional development for improved patient
Introduction
Pathologic analysis of tumour tissue is the gold standard for establishing a cancer diagnosis. Following the histological diagnosis of an advanced or early malignancy, genomic analysis is often necessary to guide treatment selection and aid prognosis. However, the requirement for sufficient tissue for genomic testing can lead to repeat invasive biopsies and subsequent delays in initiating therapy. In some cases, repeat biopsy is not feasible for many reasons, such as difficult tumour location, procedural availability and patient preference[1].
Cell-free DNA (cfDNA) refers to extracellular DNA molecules that originate from any cell type found in body fluids. Fragments of DNA are released into circulation as cfDNA by active release or passively through apoptosis and necrosis[2]. The fraction of cfDNA that originates from tumour cells is known as circulating tumour DNA (ctDNA). Although ctDNA can be measured in several bodily fluids, including urine, saliva, pleural and cerebrospinal fluid, it is most commonly assessed in plasma[3]. In this article, we discuss the current and future uses of ctDNA in guiding cancer treatments and the role of tumour molecular boards in interpreting these data.
Methods of ctDNA detection
Several technologies are used for ctDNA detection in plasma, primarily polymerase chain reaction (PCR) and next generation sequencing (NGS). PCR-based approaches include digital droplet PCR (ddPCR) and BEAMing (beads, emulsion, amplification, magnetics). These assays are commonly used to assess for single or few well characterised mutations in clinically relevant genes[3,4]. One example is the ‘cobas EGFR mutation test v2’, a real-time PCR diagnostic test designed to detect 42 mutations in exons 18-21 of EGFR (epidermal growth factor receptor) gene[5]. Activating alterations in EGFR occur in 10% to 20% of European and 30 to 50% of Asian non-small cell lung cancer (NSCLC) patients. This oncogene can be targeted with EGFR tyrosine kinase inhibitors (TKIs), which can yield high objective response rates and durable responses[6,7]. This assay may be employed to genotype EGFR in the assessment of a patient newly diagnosed with advanced NSCLC, or in a patient with a known EGFR mutation to search for the resistance alteration, T790M, following disease progression on a first or second generation EGFR TKI[8].
PCR-based methods are relatively inexpensive and have fast turnaround times compared to NGS; however, NGS offers several benefits over PCR, including the facilitation of broad-based genomic testing of multiple genes simultaneously. ctDNA PCR is also limited to detecting known mutations while ctDNA NGS can detect rare and unknown variants and amplifications[4]. Although PCR methods have high sensitivity, improvements in NGS has led to a variant allele frequency (VAF) lower limit of detection of 0.01-1%[2–4].
Clinical uses of ctDNA for cancer treatment
The most frequent clinical application of ctDNA in the oncology clinic is the genotyping of a newly diagnosed advanced malignancy to assess for actionable and prognostic genetic variants, because adequate tumour tissue for molecular analysis may not be available after the initial histological assessment. ctDNA is also commonly assessed in patients with progressive disease to ascertain new driver or resistance mechanisms such as detecting the aforementioned T790M resistance mutation in EGFR mutant NSCLC who have progressed on first or second-line EGFR TKIs[9,10]. Liquid biopsies are useful in these scenarios because a repeat tissue biopsy may not always be feasible[3].
ctDNA can also be assessed during treatment; reduction in ctDNA levels in plasma post treatment initiation has been shown to be associated with superior treatment outcomes in multiple tumour types[11–16]. However, serial monitoring of ctDNA quantities is not currently guideline recommended because its clinical impact has not been established, and thus it is often limited to clinical trials.
Advantages and disadvantages of ctDNA genotyping
There are many advantages to using ctDNA for genotyping. For exampIe, it is minimally invasive, requiring a simple blood draw. The turnaround time for ctDNA NGS results are considerably faster than the tissue testing pathway, as evidenced in the NILE study, where newly diagnosed NSCLC patients had plasma samples taken for NGS ctDNA analysis at the same time as standard of care tissue genotyping. The study results showed that ctDNA was non-inferior to tissue testing at detecting guideline recommended biomarkers and had a faster median turnaround time (9 versus 15 days; P<0.0001)[17].
Tumour heterogeneity refers to the genomic alterations within a cancer that arise from tumour cell evolution. Owing to intra-tumour heterogeneity, a tissue biopsy may sample an area that is not reflective of the dominant alterations of the tumour[18]. Assessing ctDNA can overcome this challenge and capture multiclonal heterogeneity. In 2021, Lim et al. assessed ctDNA in 93 patients with advanced colorectal cancer who were found to be RAS wildtype on tumour tissue analysis. Investigational ctDNA assessment found KRAS/NRAS mutations in 8% of patients who later had poor responses to cetuximab-containing chemotherapy[15]. This study highlights how ctDNA sequencing can be used to provide additional information to better reflect tumour mutation status and predict treatment outcomes[15].
Disadvantages of ctDNA include its inferiority to tissue NGS at detecting gene fusions and copy number variations, which may lead to false negative results[3,4,19,20]. As the amount of tumour released into the blood increases with stage, false negatives can also occur owing to low total body tumour burden, such as in oligometastatic disease states. Patients with brain metastases may have undetectable ctDNA in their plasma. Negative ctDNA NGS should be interpreted with caution and confirmed using tissue-based testing methods if possible[4].
False positives can occur when mutations present in the DNA of white cells are shed into the blood stream, referred to as clonal haematopoiesis (CH). CH is part of the normal process of aging with the accumulation of somatic mutations and clonal expansion of hematopoietic stem cells[21]. Physicians must consider CH when interpreting a result in older patients, especially if the variant is not typical for the primary tumour subtype. Alterations in DNMT3A, TET2, ASXL1 and JAK2 are frequently seen in CH[21].
As mentioned, ctDNA quantities correlate with burden of disease, therein patients with advanced solid tumours (stage III/IV) are more likely to shed ctDNA into the circulation. As a result, ctDNA assays are not currently used to genotype early cancers because the result is unlikely to be informative, this may change in the future as assays become more sensitive[4,22].
Applications of ctDNA in common solid tumours and guideline recommendations
Breast cancer
The European Society for Medical Oncology (ESMO) guidelines recommend the assessment of ctDNA in breast cancer if the result will alter the treatment approach; for example, by detecting an activating PIK3CA alteration in ER/PR positive HER2 negative metastatic breast cancer to guide the use of alpelisib[23]. The National Comprehensive Cancer Network (NCCN) guidelines recommend the use of ctDNA assays to detect PIK3CA alterations but a tissue biopsy should be pursued if ctDNA is negative. A concordance rate of 81% has been described for the detection of PIK3CA alterations in blood and tissue of breast cancer patients[24]. Although dynamic changes in ctDNA levels have been shown to correlate with treatment outcomes in breast cancer, it is not currently guideline recommended outside of research settings[16,25].
Colorectal cancer
We have discussed that advanced colorectal tumours require genotyping to guide treatment, as mutations in the RAS (KRAS, NRAS, BRAF) genes are negative predictors of response to anti-EGFR therapy[26]. The European Medicines Agency (EMA) has approved the use of ctDNA PCR-based assays for the assessment of the presence of KRAS, NRAS and BRAF alterations in colorectal cancer. Despite their high concordance rates, these assays are currently not recommended by international guidelines[27]. The ESMO guidelines note that the utility of liquid biopsies to guide treatment in colorectal cancer is still being investigated, and cannot yet be recommended in routine practice[26]. Like other disease states, assessing ctDNA or minimal residual disease post operatively has been studied in the adjuvant colorectal cancer setting. In 2019, a study of 100 patients led Tie et al. to demonstrate that serially assessing ctDNA post-operatively and post- adjuvant chemotherapy could provide a real-time indication of adjuvant therapy efficacy in stage III colon cancer[28]. More recently, presented at the 2022 American Society of Clinical Oncology Congress, the DYNAMIC study demonstrated that ctDNA could be used to guide adjuvant chemotherapy in stage II colon cancer[29]. Nonetheless large prospective data are lacking and ctDNA analysis is currently not recommended as standard care in colorectal cancer.
Ovarian cancer
Comparable to other malignancies, ctDNA concentrations are correlated with stages of high-grade serous ovarian cancer (HGSOC) and ctDNA fluctuations may reflect response to treatment[30]. TP53 mutations are present in 95% of HGSOC and changes in ctDNA allelic frequencies of TP53 variants have been investigated as a biomarker of treatment response. In 2016, Parkinson et al. found that a decrease of ≤60% in TP53 plasma mutant allele fraction after one cycle of chemotherapy was associated with shorter time to progression[30].
Knowledge of the germline/somatic BRCA status and homologous recombination deficiency (HRD) status of HGSOC is pertinent to guide and predict response to poly ADP-ribose polymerase (PARP) inhibitor treatment[31]. A key resistance mechanism to platinum-based chemotherapies and PARP inhibitors in BRCA-mutant cancers is the acquisition of BRCA reversion mutations that restore protein function. BRCA reversion mutations can be detected in ctDNA prior to and during treatment and are associated with decreased clinical benefit to PARP inhibitor therapy[12]. Such is an example of tumour clonal evolution captured on ctDNA. Routine analysis of ctDNA is not currently recommended by international guidelines in the management of HGSOC but is expected in the future[32,33].
Prostate cancer
Alterations in the homologous recombination repair (HRR) genes (e.g. BRCA1, BRCA2, ATM) are predictive of the clinical benefit of PARP inhibitors in prostate cancer[34]. The PROfound study demonstrated the efficacy of olaparib in patients with metastatic castrate resistant prostate cancer (mCRPC) with germline or somatic HRR alterations[34]. Analysis of the ctDNA of a subset of the PROfound study population found that ctDNA was concordant with tissue results in 80% of cases[35]. The NCCN guidelines recommend that any clinically validated ctDNA test can be used to identify mCRPC patients for PARP inhibitor treatment (olaparib or rucaparib), but tissue testing is preferred. The current ESMO guidelines have no formal recommendations for plasma-based testing in prostate cancer[36,37].
Non-small cell lung cancer
Owing to the ever-expanding understanding of targetable alterations in advanced NSCLC, broad-based genomic testing performed by NGS is the preferred method of NSCLC genotyping, enabling multiple genomic alterations to be assessed simultaneously[38]. International guidelines and the NHS England genomic test directory advise assessment of EGFR, ALK, ROS1, RET, METex14, BRAFV600E, KRAG12C, and NTRK1-3 in advanced NSCLC[27,38]. As discussed, high concordance between plasma ctDNA analysis with tissue profiling has been demonstrated in NSCLC[17]. The NCCN guidelines state that ctDNA should not replace tissue biopsy in NSCLC but can be used when a patient is not medically fit for invasive tissue sampling or there is insufficient tissue for molecular analysis[39]. ESMO guidelines do not currently address using ctDNA to genotype newly diagnosed NSCLC. Both guidelines recommend the use of ctDNA to detect resistance mechanisms in EGFR mutant NSCLC, because such resistance mechanisms may be targetable by another agent or facilitate enrolment into clinical trials, such as C797S, which is a common resistance mechanism to first-line osimertinib[7,40]. If a ctDNA test is negative in such cases, a tissue biopsy should be pursued. Clinicians should be aware that a liquid biopsy is less sensitive at detecting MET or ERBB2 amplifications, which may be an off-target mechanism of resistance in EGFR mutant NSCLC. Furthermore, only tissue sampling can identify small cell transformation, which occurs in 5% of progressive mutant EGFR NSCLC[7,41]. International guidelines note that prognostic value may be derived from serial ctDNA testing during treatment, but it is not currently recommended as part of routine care.
In 2021, the International Association for the Study of Lung Cancer (IASLC) issued a consensus statement regarding the use of liquid biopsy in advanced NSCLC[4]. This describes three potential pathways for using ctDNA to genotype a newly diagnosed NSLCC. Like ESMO and NCCN, it describes the sequential approach, where ctDNA can be used as a salvage pathway following the failure of tissue analysis. It also considers the complementary approach; where ctDNA can be used at the same time as tissue genotyping as it may provide clinicians with extra genomic information. The statement also describes the ‘plasma-first approach’ — when tissue is unavailable for testing proceed with ctDNA and consider re-biopsy if no targetable alterations are found in plasma[4].
The role of the molecular tumour board
With the increasing number of oncology drug targets and genetic testing platforms, resources are required to aid physicians in understanding and utilising molecular reports. This led to the development of the molecular tumour board (MTB). The purpose of an MTB is to interpret tumour molecular reports and make genomically matched treatment recommendations based on the results and clinical details. The MTB uses a patient-centred approach and members traditionally include oncologists, clinical geneticists, pathologists and bioinformaticians. More recently, clinical nurse specialists, genetics counsellors and pharmacists have also become important members of the MTB team[42,43]. A recommendation may include an unlicensed medication, a clinical trial or the MTB may decide further testing is required[42].
Several oncology precision medicine databases are available to assist MTBs and physicians in deciphering a genomic report and to determine if a variant is clinically actionable. A popular tool is OncoKB, developed by the Memorial Sloan Kettering Cancer Centre — a decision support tool that contains biological and clinical information regarding somatic cancer variants[44]. On entering a variant into the database, users are supplied with information regarding the alteration’s oncogenicity and clinical actionability[44].
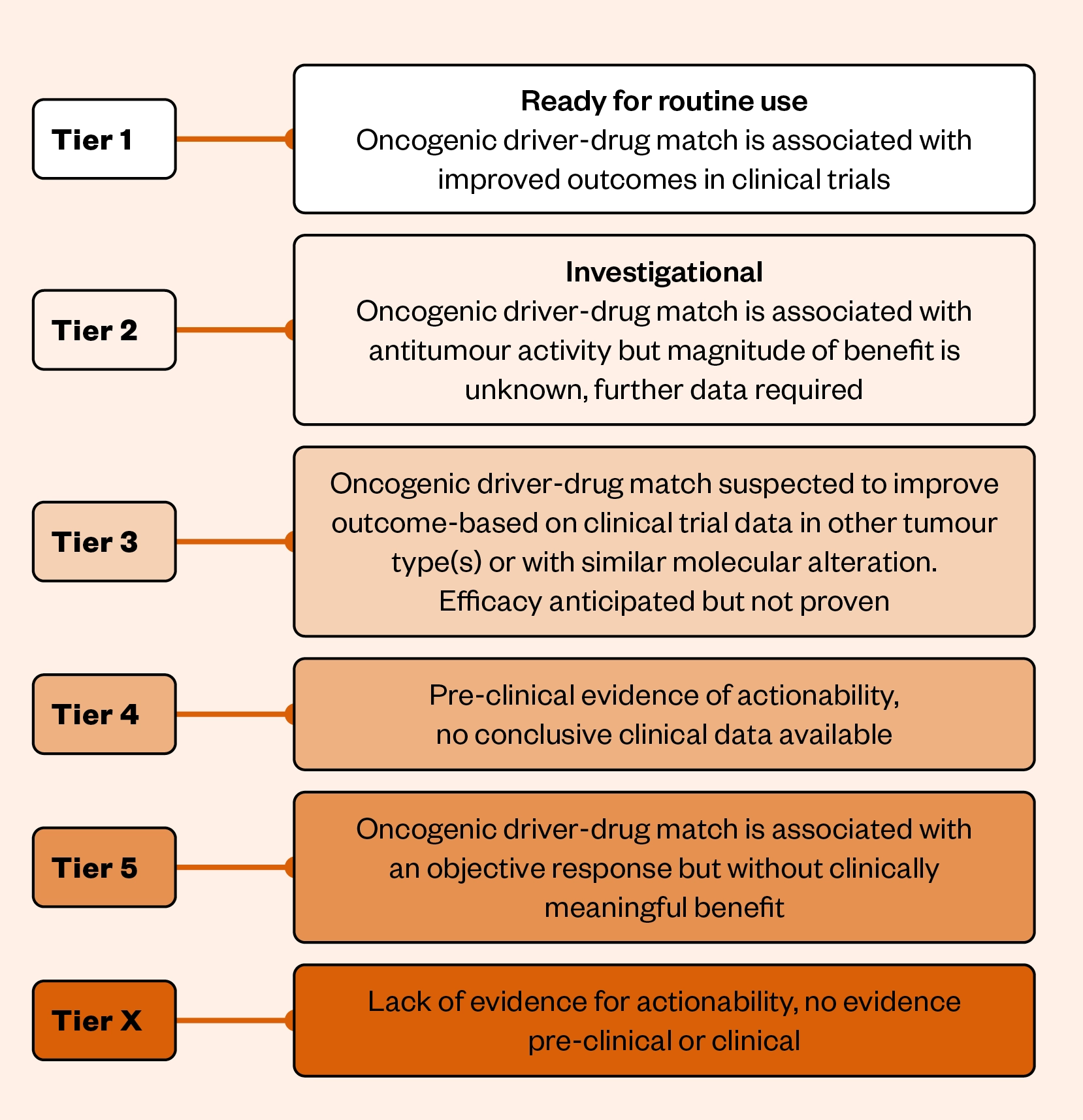
ESMO has created a Scale for Clinical Actionability of Molecular Targets, known as ‘ESCAT’[45]. ESCAT classifies mutations into tiers to develop common terminology to support treatment decisions (see Figure[45]). The classification has six tiers in total with tier 1 targets having the highest level of supporting evidence and tier X the lowest.
An example of a tier 1 target includes an ALK fusion in NSCLC treated with lorlatinib, which has a proven survival benefit as demonstrated by the CROWN phase III clinical trial[46]. Tier 2 targets fall into the investigational category, in which both retrospective and prospective studies demonstrate clinical activity. However, the magnitude of benefit in terms of survival endpoints remains under investigation, for example the use of trastuzumab and pertuzumab to treat ERBB2/HER2 amplified colorectal cancer[47].
Within an MTB, as with any multidisciplinary approach, pharmacists add valuable input as professionals with expertise in pharmacology, drug interactions and experience of applying evidence-based practice alongside in-depth knowledge of commissioning pathways to support therapeutic decision making. Pharmacists can support individual funding requests and work with drugs and therapeutic committees to critically appraise clinical outcomes versus treatment costs when a genomic alteration is linked to a non-commissioned treatment, such as ERBB2/HER2 mutations in NSCLC, for which there are growing data that the antibody drug conjugate trastuzumab deruxtecan is active but to date it is only approved by the EMA for metastatic HER2-positive breast cancer that has received one or more prior anti HER2-based regimens[48,49].
Table 1 describes common targets in oncology, their ESMO-ESCAT tiers, and the availability of associated treatments within the NHS[45,50,51].
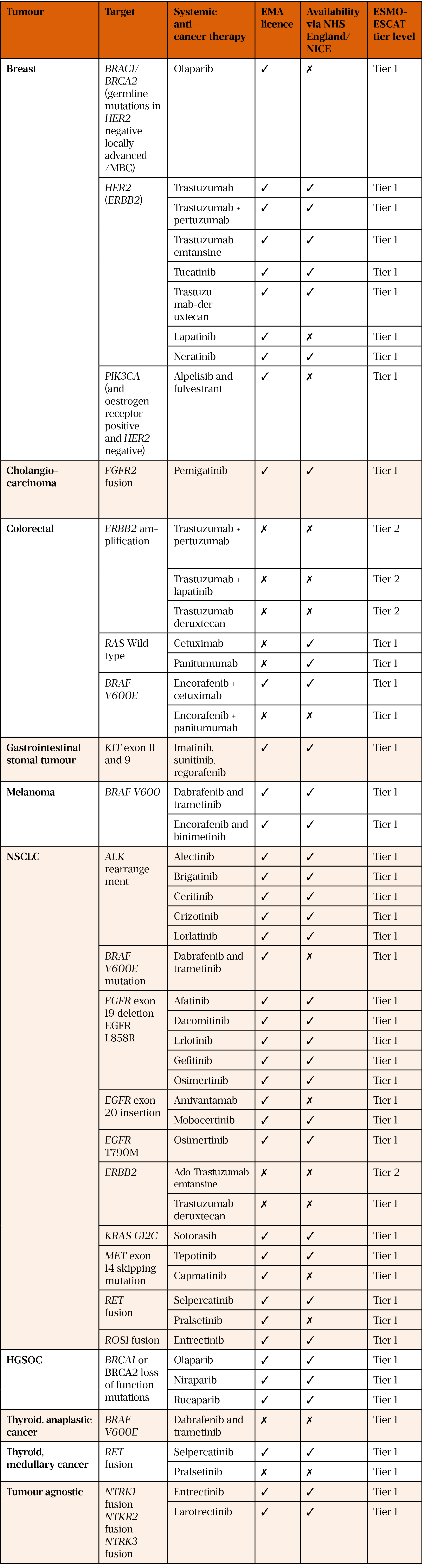
EMA: European Medicines Agency; ESMO: European Society for Medical Oncology;
ESMO-ESCAT: ESMO scale for clinical actionability of molecular targets; HGSOC: high-grade
serious ovarian cancer; NICE: National Institute for Health and Care Excellence; NSCLC: non-small cell lung cancer
The MTB provides a platform for improving genomic literacy and developing clear reporting standards between clinicians and clinical scientists, particularly as genomic technology and cancer drug development is ever evolving. It provides a clear interpretation and summary of outcomes by indicating if a genomic aberration is truly targetable or of biological interest only, such as TP53 and RB1 mutations in EGFR mutant NSCLC. These alterations are not currently targetable, but are associated with poorer outcomes[7,52,53].
Interpretation of a ctDNA result
When interpreting a ctDNA result, like any investigation, one should note what technology was used; a PCR assay focusing on a few mutations of a single gene, or a broad multigene NGS-based test. If interested in variants of a particular gene, the MTB will check if the panel in use assessed the gene and necessary region in question. A multigene NGS panel may only analyse part of a gene, hence a negative result only indicates an absence of a mutation in the regions tested[3,54].
The previously described ctDNA limitations must also be considered when interpreting a result. If a negative result is found, it may mean no alterations are present in the genes tested or insufficient tumour DNA being shed into the circulation. In such cases, a tissue biopsy should be considered if clinically necessary[4].
When a variant is detected, the MTB will consider if it is tumour derived, germline (heritable), or related to CH; the VAF can aid in the interpretation of the result. VAFs, reported as percentages, describe the number of variant mutant reads over the total number of reads in a sample. As with tumour tissue sequencing, incidental germline pathogenic variants can be detected[55]. A heterozygous mutation of germline origin would be expected to be present in 50% of reads: one wildtype allele and one mutant allele. A VAF near 50% suggests a possible germline variant and referral to clinical genetics should be considered[55]. As described, ctDNA can detect alterations secondary to CH and this should be considered when interpreting the results of older patients[21].
The MTB and decision support tools can help decide the clinical impact of the variants detected. The decision will be guided by the variant tier, tumour type, stage, patient performance status and prior therapies[53].
Future applications of ctDNA
Tumour mutational burden (TMB), defined as the total number of somatic mutations per coding area of a tumour genome, is a biomarker to predict response to immune check point inhibitors (ICIs)[56]. Studies have shown that high TMB, assessed via ctDNA in blood (bTMB), is associated with improved progression-free survival and response rates with ICI[57,58]. bTMB may be used as an alternative surrogate for tissue TMB testing in the future.
Another exciting area of growth in the application of ctDNA technology is in the surveillance of radically treated cancer patients. Minimal residual disease (MRD) is a term that describes the presence of ctDNA after the initiation of curative treatment, with no evidence of disease radiologically[59]. The presence of detectable ctDNA represents the existence of occult micro metastatic disease. Studies have shown that the detection of ctDNA (i.e. MRD) in patients following the initiation of radical therapy is associated with inferior recurrence-free survival, when compared with patients without detectable residual disease[59–62]. Large prospective trials are ongoing randomising patients to further treatment based on the presence of ctDNA MRD post radical treatment, such as the aforementioned DYNAMIC study in colorectal cancer[29]. The aim of ctDNA MRD analysis is to allow personalisation of adjuvant therapy to increase the likelihood of cure and reduce treatment-related toxicity. It has not yet been determined in many cancer types whether escalating treatment based on MRD status will alter the natural history of a patient’s cancer and improve survival[3].
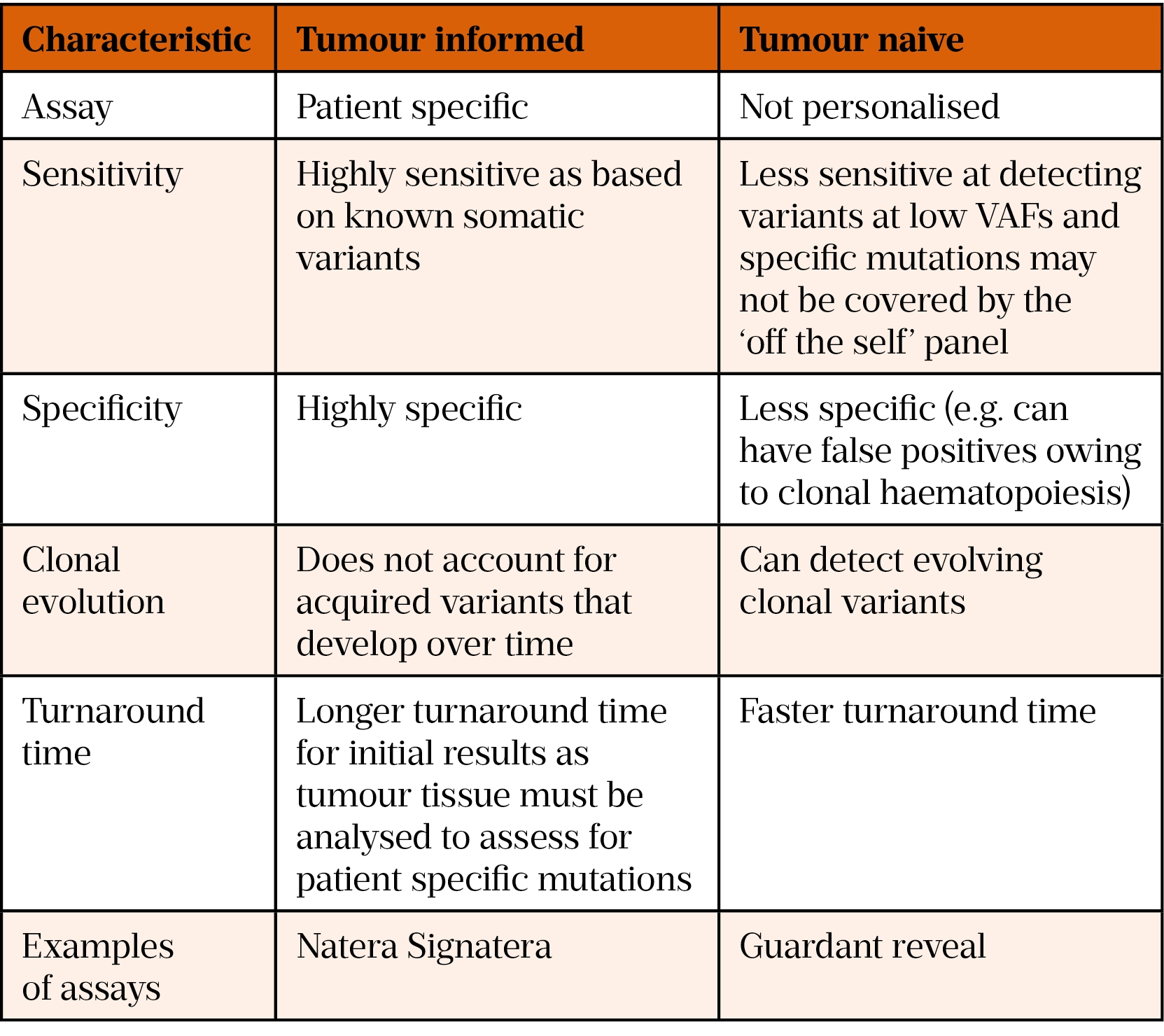
Methods to assess MRD can be divided into tumour genotype-informed and tumour genotype-naïve. The tumour genotype-informed approach involves analysis of a patient’s tumour and plasma; a personalised panel is developed to detect patient-specific mutations, including SNVs, indels, fusions and CNAs, identified through analysis of the tumour DNA[59]. This panel is unique to each patient and is used to analyse serial plasma specimens to track these specific mutations. In contrast, tumour genotype-naïve methods employ panels designed to cover genes recurrently mutated in the subtype of cancer and are not patient specific[59]. Table 2 describes the differences between tumour informed and tumour naïve assays[63,64].
The incorporation of ctDNA fragment length analysis in ctDNA assays is undergoing development to improve sensitivity, as the length of ctDNA is shorter than that of cfDNA of healthy cells[65]. This technology has been employed routinely in non-invasive prenatal testing for years, owing to the fragment length of foetal cfDNA being shorter than that of maternal cfDNA[66]. The DELFI assay (DNA evaluation of fragments for early interception) uses a machine learning model incorporating genome-wide fragmentation. This assay is under investigation as a cancer screening tool. The genome-wide pattern from an individual can be compared to reference populations to determine if the fragmentation pattern is likely benign or cancer-derived[67].
Methylation ctDNA assays are also under investigation in cancer screening. This includes the GRAIL assay, which measures aberrantly methylated DNA. Methylation alterations are more common in plasma than mutant DNA, and validation studies have demonstrated the efficacy of this assay at predicting the cancer signal origin[68,69]. It is predicted that methylation based ctDNA assays will be used as a complement to existing single-cancer screening tests in the future.
Conclusion
ctDNA assays are a valuable tool for the detection of predictive biomarkers to guide treatment in oncology, and in the future will likely be incorporated in the diagnosis, surveillance, and screening of patients. As the complexity of cancer biomarkers continues to grow and systemic anti-cancer therapy moves further toward targeted agents, large genomic panels will be more commonly run on ctDNA. This underlines the importance of rapidly educating clinicians and other healthcare professionals on the applications and limitations of ctDNA to ensure high-quality patient care.
- 1Lung Cancer Diagnosis and Management. National Institute for Health and Care Excellence. https://www.nice.org.uk/guidance/ng122 (accessed Sep 2022).
- 2Merker JD, Oxnard GR, Compton C, et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. JCO. 2018;36:1631–41. doi:10.1200/jco.2017.76.8671
- 3Pascual J, Attard G, Bidard F-C, et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO Precision Medicine Working Group. Annals of Oncology. 2022;33:750–68. doi:10.1016/j.annonc.2022.05.520
- 4Rolfo C, Mack P, Scagliotti GV, et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. Journal of Thoracic Oncology. 2021;16:1647–62. doi:10.1016/j.jtho.2021.06.017
- 5Tie J, Wang Y, Cohen J, et al. Circulating tumor DNA dynamics and recurrence risk in patients undergoing curative intent resection of colorectal cancer liver metastases: A prospective cohort study. PLoS Med. 2021;18:e1003620. doi:10.1371/journal.pmed.1003620
- 6Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N Engl J Med. 2020;382:41–50. doi:10.1056/nejmoa1913662
- 7Passaro A, Leighl N, Blackhall F, et al. ESMO expert consensus statements on the management of EGFR mutant non-small-cell lung cancer. Annals of Oncology. 2022;33:466–87. doi:10.1016/j.annonc.2022.02.003
- 8Rosell R, Moran T, Queralt C, et al. Screening for Epidermal Growth Factor Receptor Mutations in Lung Cancer. N Engl J Med. 2009;361:958–67. doi:10.1056/nejmoa0904554
- 9Mok TS, Wu Y-L, Ahn M-J, et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N Engl J Med. 2017;376:629–40. doi:10.1056/nejmoa1612674
- 10Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473–81. doi:10.1038/nrclinonc.2014.104
- 11Soo RA, Martini J-F, van der Wekken AJ, et al. Early circulating tumor (ct) DNA dynamics and efficacy of lorlatinib: Analysis from the CROWN study. JCO. 2021;39:9011–9011. doi:10.1200/jco.2021.39.15_suppl.9011
- 12Gray JE, Okamoto I, Sriuranpong V, et al. Tissue and Plasma EGFR Mutation Analysis in the FLAURA Trial: Osimertinib versus Comparator EGFR Tyrosine Kinase Inhibitor as First-Line Treatment in Patients with EGFR-Mutated Advanced Non–Small Cell Lung Cancer. Clinical Cancer Research. 2019;25:6644–52. doi:10.1158/1078-0432.ccr-19-1126
- 13Weber S, van der Leest P, Donker HC, et al. Dynamic Changes of Circulating Tumor DNA Predict Clinical Outcome in Patients With Advanced Non–Small-Cell Lung Cancer Treated With Immune Checkpoint Inhibitors. JCO Precision Oncology. 2021;:1540–53. doi:10.1200/po.21.00182
- 14Wang H, Zhou F, Qiao M, et al. The Role of Circulating Tumor DNA in Advanced Non-Small Cell Lung Cancer Patients Treated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Front. Oncol. 2021;11. doi:10.3389/fonc.2021.671874
- 15Lim Y, Kim S, Kang J-K, et al. Circulating tumor DNA sequencing in colorectal cancer patients treated with first-line chemotherapy with anti-EGFR. Sci Rep. 2021;11. doi:10.1038/s41598-021-95345-4
- 16Hrebien S, Citi V, Garcia-Murillas I, et al. Early ctDNA dynamics as a surrogate for progression-free survival in advanced breast cancer in the BEECH trial. Annals of Oncology. 2019;30:945–52. doi:10.1093/annonc/mdz085
- 17Leighl NB, Page RD, Raymond VM, et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non–small Cell Lung Cancer. Clinical Cancer Research. 2019;25:4691–700. doi:10.1158/1078-0432.ccr-19-0624
- 18Yang M, Topaloglu U, Petty WJ, et al. Circulating mutational portrait of cancer: manifestation of aggressive clonal events in both early and late stages. J Hematol Oncol. 2017;10. doi:10.1186/s13045-017-0468-1
- 19Schrock AB, Welsh A, Chung JH, et al. Hybrid Capture–Based Genomic Profiling of Circulating Tumor DNA from Patients with Advanced Non–Small Cell Lung Cancer. Journal of Thoracic Oncology. 2019;14:255–64. doi:10.1016/j.jtho.2018.10.008
- 20Rolfo C, Mack PC, Scagliotti GV, et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. Journal of Thoracic Oncology. 2018;13:1248–68. doi:10.1016/j.jtho.2018.05.030
- 21Chan HT, Chin YM, Nakamura Y, et al. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers. 2020;12:2277. doi:10.3390/cancers12082277
- 22Rolfo C, Russo A. Liquid biopsy for early stage lung cancer moves ever closer. Nat Rev Clin Oncol. 2020;17:523–4. doi:10.1038/s41571-020-0393-z
- 23Gennari A, André F, Barrios CH, et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Annals of Oncology. 2021;32:1475–95. doi:10.1016/j.annonc.2021.09.019
- 24Davis AA, Jacob S, Gerratana L, et al. Landscape of circulating tumour DNA in metastatic breast cancer. EBioMedicine. 2020;58:102914. doi:10.1016/j.ebiom.2020.102914
- 25Moding EJ, Liu Y, Nabet BY, et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nat Cancer. 2020;1:176–83. doi:10.1038/s43018-019-0011-0
- 26Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Annals of Oncology. 2016;27:1386–422. doi:10.1093/annonc/mdw235
- 27Pinheiro M, Peixoto A, Rocha P, et al. KRAS and NRAS mutational analysis in plasma ctDNA from patients with metastatic colorectal cancer by real-time PCR and digital PCR. Int J Colorectal Dis. 2022;37:895–905. doi:10.1007/s00384-022-04126-6
- 28Tie J, Cohen JD, Wang Y, et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019;5:1710. doi:10.1001/jamaoncol.2019.3616
- 29Tie J, Cohen JD, Lahouel K, et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N Engl J Med. 2022;386:2261–72. doi:10.1056/nejmoa2200075
- 30Parkinson CA, Gale D, Piskorz AM, et al. Exploratory Analysis of TP53 Mutations in Circulating Tumour DNA as Biomarkers of Treatment Response for Patients with Relapsed High-Grade Serous Ovarian Carcinoma: A Retrospective Study. PLoS Med. 2016;13:e1002198. doi:10.1371/journal.pmed.1002198
- 31NCCN Guidelines: Ovarian Cancer. National Comprehensive Cancer Network . https://www.nccn.org/login?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf (accessed Sep 2022).
- 32Colombo N, Sessa C, du Bois A, et al. ESMO–ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Annals of Oncology. 2019;30:672–705. doi:10.1093/annonc/mdz062
- 33Chabon JJ, Hamilton EG, Kurtz DM, et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–51. doi:10.1038/s41586-020-2140-0
- 34Hussain M, Mateo J, Fizazi K, et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2020;383:2345–57. doi:10.1056/nejmoa2022485
- 35Chi KN, Barnicle A, Sibilla C, et al. Concordance of BRCA1, BRCA2 (BRCA), and ATM mutations identified in matched tumor tissue and circulating tumor DNA (ctDNA) in men with metastatic castration-resistant prostate cancer (mCRPC) screened in the PROfound study. JCO. 2021;39:26–26. doi:10.1200/jco.2021.39.6_suppl.26
- 36Parker C, Castro E, Fizazi K, et al. Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2020;31:1119–34. doi:10.1016/j.annonc.2020.06.011
- 37Abida W, Patnaik A, Campbell D, et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. JCO. 2020;38:3763–72. doi:10.1200/jco.20.01035
- 38Planchard D, Popat S, Kerr K, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2018;29:iv192–237. doi:10.1093/annonc/mdy275
- 39NCCN Guidelines: NSCLC. National Comprehensive Cancer Network . https://www.nccn.org/login?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed Sep 2022).
- 40Zhao H-Y, Xi X-X, Xin M, et al. Overcoming C797S mutation: The challenges and prospects of the fourth-generation EGFR-TKIs. Bioorganic Chemistry. 2022;128:106057. doi:10.1016/j.bioorg.2022.106057
- 41Oser MG, Niederst MJ, Sequist LV, et al. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. The Lancet Oncology. 2015;16:e165–72. doi:10.1016/s1470-2045(14)71180-5
- 42van der Velden DL, van Herpen CML, van Laarhoven HWM, et al. Molecular Tumor Boards: current practice and future needs. Annals of Oncology. 2017;28:3070–5. doi:10.1093/annonc/mdx528
- 43Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized Oncology Through Integrative High-Throughput Sequencing: A Pilot Study. Sci. Transl. Med. 2011;3. doi:10.1126/scitranslmed.3003161
- 44Chakravarty D, Gao J, Phillips S, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precision Oncology. 2017;:1–16. doi:10.1200/po.17.00011
- 45Mateo J, Chakravarty D, Dienstmann R, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Annals of Oncology. 2018;29:1895–902. doi:10.1093/annonc/mdy263
- 46Shaw AT, Bauer TM, de Marinis F, et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N Engl J Med. 2020;383:2018–29. doi:10.1056/nejmoa2027187
- 47Meric-Bernstam F, Hurwitz H, Raghav KPS, et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. The Lancet Oncology. 2019;20:518–30. doi:10.1016/s1470-2045(18)30904-5
- 48Li BT, Smit EF, Goto Y, et al. Trastuzumab Deruxtecan in HER2-Mutant Non–Small-Cell Lung Cancer. N Engl J Med. 2022;386:241–51. doi:10.1056/nejmoa2112431
- 49Enhertu. European Medicines Agency. https://www.ema.europa.eu/en/medicines/human/EPAR/enhertu (accessed Sep 2022).
- 50Therapeutic Areas Latest Updates: Cancer. European Medicines Agency. https://www.ema.europa.eu/en/news-events/therapeutic-areas-latest-updates/cancer (accessed Sep 2022).
- 51Cancer Drugs Fund List. NHS England. https://www.england.nhs.uk/cancer/cdf/cancer-drugs-fund-list/ (accessed Sep 2022).
- 52Huang B, Chen Q, Allison D, et al. Molecular Tumor Board Review and Improved Overall Survival in Non–Small-Cell Lung Cancer. JCO Precision Oncology. 2021;:1530–9. doi:10.1200/po.21.00210
- 53Luchini C, Lawlor RT, Milella M, et al. Molecular Tumor Boards in Clinical Practice. Trends in Cancer. 2020;6:738–44. doi:10.1016/j.trecan.2020.05.008
- 54Mateo J, Steuten L, Aftimos P, et al. Delivering precision oncology to patients with cancer. Nat Med. 2022;28:658–65. doi:10.1038/s41591-022-01717-2
- 55Mandelker D, Donoghue M, Talukdar S, et al. Germline-focussed analysis of tumour-only sequencing: recommendations from the ESMO Precision Medicine Working Group. Annals of Oncology. 2019;30:1221–31. doi:10.1093/annonc/mdz136
- 56Fusco MJ, West H (Jack), Walko CM. Tumor Mutation Burden and Cancer Treatment. JAMA Oncol. 2021;7:316. doi:10.1001/jamaoncol.2020.6371
- 57Rizvi NA, Cho BC, Reinmuth N, et al. Durvalumab With or Without Tremelimumab vs Standard Chemotherapy in First-line Treatment of Metastatic Non–Small Cell Lung Cancer. JAMA Oncol. 2020;6:661. doi:10.1001/jamaoncol.2020.0237
- 58Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24:1441–8. doi:10.1038/s41591-018-0134-3
- 59Moding EJ, Nabet BY, Alizadeh AA, et al. Detecting Liquid Remnants of Solid Tumors: Circulating Tumor DNA Minimal Residual Disease. Cancer Discovery. 2021;11:2968–86. doi:10.1158/2159-8290.cd-21-0634
- 60Chen K, Zhao H, Shi Y, et al. Perioperative Dynamic Changes in Circulating Tumor DNA in Patients with Lung Cancer (DYNAMIC). Clinical Cancer Research. 2019;25:7058–67. doi:10.1158/1078-0432.ccr-19-1213
- 61Abbosh C, Birkbak NJ, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–51. doi:10.1038/nature22364
- 62Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discovery. 2017;7:1394–403. doi:10.1158/2159-8290.cd-17-0716
- 63Natera (2019) Signatera: Seeing beyond the limit: Detect residual disease and assess treatment respons. Natera. https://www.natera.com/wp-content/uploads/2020/11/Oncology-Clinical-Seeing-beyond-the-limit-Detect-residual-disease-and-assess-treatment-response-SGN_AV_WP.pdf (accessed Sep 2022).
- 64Guardant Guardant RevealTM liquid biopsy test. Guardant Health. https://guardanthealth.com/wp-content/uploads/Backgrounder-Guardant-Reveal-1.pdf
- 65Underhill HR. Leveraging the Fragment Length of Circulating Tumour DNA to Improve Molecular Profiling of Solid Tumour Malignancies with Next-Generation Sequencing: A Pathway to Advanced Non-invasive Diagnostics in Precision Oncology? Mol Diagn Ther. 2021;25:389–408. doi:10.1007/s40291-021-00534-6
- 66Kohabir K, Wolthuis R, Sistermans EA. Fragmentomic cfDNA Patterns in Noninvasive Prenatal Testing and Beyond. J. Biomed. Transl. Res. 2021;7:38–47. doi:10.14710/jbtr.v7i1.10229
- 67Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–9. doi:10.1038/s41586-019-1272-6
- 68Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Annals of Oncology. 2020;31:745–59. doi:10.1016/j.annonc.2020.02.011
- 69Klein EA, Richards D, Cohn A, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Annals of Oncology. 2021;32:1167–77. doi:10.1016/j.annonc.2021.05.806