Abstract
Diverse insulin preparations are available for use in people with diabetes in the UK and, to some extent, can be matched to the wide range of clinical scenarios in which they are used. Insulin is a peptide and therefore a biological medicine, and has undergone a cascade of ‘biobetter’ development over 100 years, from purification to manufacturing to altered absorption properties, alongside developments in delivery devices and their interaction with blood glucose-monitoring techniques. Meanwhile, an advancing choice of both mealtime and basal insulin analogue preparations has been marketed with controversial advantages, such as limited or no advantage to the primary measure of glucose control — HbA1c — and usually at increased cost. Further developments are on the near and longer horizons. Biosimilar versions of some mealtime and basal analogues have become available in recent years, with modest cost savings, but clinical questions remain over substitution and interchangeability with originator products.
Key words: insulin therapy, biosimilars, biologicals, biobetters, diabetes mellitus
Key points
- Insulin medicines have evolved over 100 years through purification technology, means of extending duration of action, recombinant DNA manufacturing technology and designer molecules;
- In type 1 diabetes mellitus, glucose control remains generally suboptimal, generating momentum for switching from a standard mealtime analogue + basal analogue pre-filled pen-injector regimen to insulin pump therapy (utilising mealtime analogues alone), to closed loop systems employing pumps controlled by continuous glucose monitoring devices. This comes at considerable cost, which often must be met from within existing diabetes care budgets;
- In type 2 diabetes mellitus, human-sequence neutral protamine Hagedorn (NPH) insulin is effective if properly used as a starter basal insulin, but has a higher risk of hypoglycaemia than insulins glargine 100 U/mL and detemir, which are in turn bettered by insulins glargine 300 U/mL and degludec;
- As a first injectable, glucagon-like peptide 1 receptor agonists (e.g. dulaglutide, liraglutide) give as good blood glucose control as basal insulin, without causing hypoglycaemia and body weight gain, and is therefore often the first-choice injectable despite higher acquisition costs. Combination therapy with insulin is possible;
- Evidence-based guidelines, which were updated most recently in 2022, are available from the National Institute for Health and Care Excellence (NICE) for ambulatory type 1 and 2 diabetes mellitus, while the Scottish Medicines Consortium endorses most of the available insulins for prescription;
- Use of insulin in hospital inpatients is not covered by NICE guidelines in some clinical scenarios, such as those with hepatic cirrhosis or renal failure, and with steroid therapy;
- Biosimilar insulins aspart, lispro and glargine 100 U/mL may offer a limited (-25%) or no cost advantage compared to originator molecules, but insulin brand substitution requires increased glucose-monitoring and diabetes care team support, which negates some of the cost advantage.
Introduction
Insulin was first discovered in 1921[1]. Although insulin has often been central in biotechnology development, essentially the same molecule is given today subcutaneously, which was not uncommon for medicines in the 1920s[2,3]. Insulin was defined as a peptide in the late 1920s, explaining its non-suitability for oral administration and its definition as a biological medicine[2].
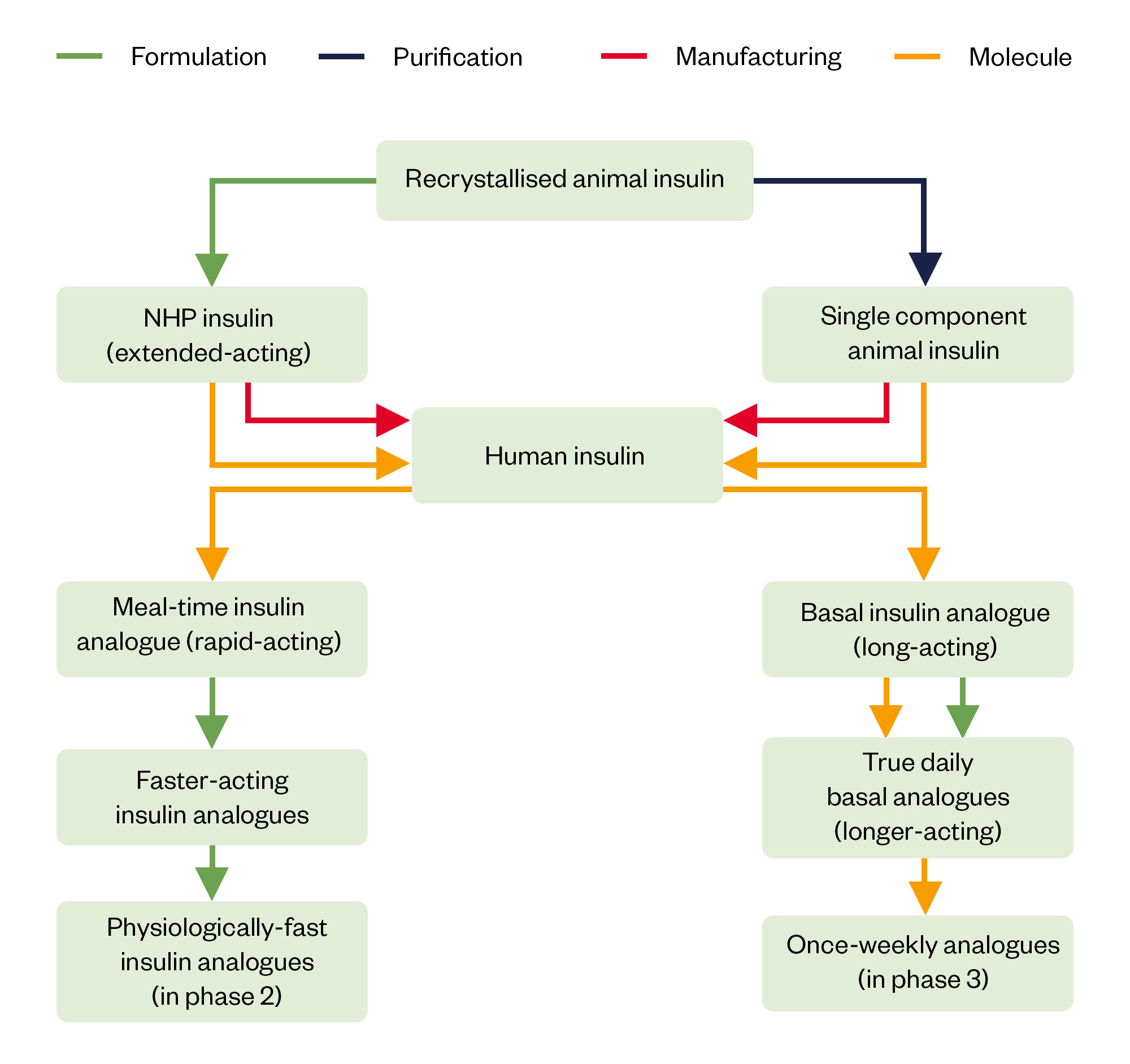
The initial preparations provided by Banting and Best had low insulin and high impurity content, setting the scene for technological improvements in insulin preparations, which continue to the current day. Recrystallisation — a serendipitous discovery by an Eli Lilly industrial biochemist — came first, but even by the early 1930s, crystals of sufficient purity to provide X-ray diffraction images were available. It took until 1948 for the first satisfactory extended-acting insulin to be available — the neutral protamine Hagedorn (NPH) insulin we still use today[2,3]. This was the first step in the cascade of biobetter insulins, where earlier clinical insulins are superseded by preparations and formulations with advantages in efficacy or tolerability. This cascade is discussed below and has become steadily more sophisticated through progress in science and biotechnology.
One major step in that process was the application of recombinant DNA technology to insulin manufacturing, technology that has enabled ‘designer’ insulins and did so by 1982. Formulation changes of excipients has also given us insulins of novel advantage and continues to do so in the 2020s[4].
Developments in insulin production have narrowed the manufacturing base, a development traceable to the 1970s, when industrial-scale column purification techniques were implemented. That was quickly followed by the move to production by fermentation in yeast and bacteria, rather than pancreatic extraction, but this sophisticated technology has limited production of quality products to three companies globally.
In the past five years, patents have expired on the first designer insulins (e.g. lispro, glulisine, and aspart), enabling the development of the insulin biosimilar market[5]. But this has largely led to the three major manufacturers (i.e. Eli Lilly, Novo Nordisk and Sanofi-Aventis) cannibalising each other, and only one other company has as yet gained market approval for its biosimilar insulin (see ‘Biosimilar insulin use’). However, the biosimilar market will become increasingly important in markets with restricted resources and other entries, such as China. This article will discuss issues around use of biosimilar insulins, which differ from use of generics and biosimilars, such as erythropoietin or anti-TNF-α medicines (e.g. etanercept, infliximab)[6,7].
Support for insulin therapy
Insulin is not a stand-alone therapy, nor is it a ‘prescribe, administer and forget’ therapy in the way that some glucose-lowering tablets can be. The reasons for this are namely that insulin requirements vary from person to person in unpredictable ways, dependent on stresses, other illnesses and disease progression. Further, the major tolerability issue — hypoglycaemia — is erratic and can be unpredictable, and it is not safe to stop insulin or make large dose reductions, which can be fatal if keto-acidosis occurs[8]. Insulin dose adjustment needs to be informed, owing to the risks of hypoglycaemia, and the possible further consequences of this in everyday life, such as when driving and operating machinery.
The National Institute for Health and Care Excellence (NICE) guidelines insist on provision of a structured education programme for anyone starting insulin, as well as educational review annually[9,10]. In practice, this extends to matters such as what to do in the event of sickness and advice for travellers (notably between time zones), to ensure adequate insulin administration, but should also provide the basis for self-dose adjustment in the majority.
Self-dose adjustment needs to be informed, not only by lifestyle events and experience of hypoglycaemia, but also by self-measurement of blood glucose levels. Usually, all people using insulin are prescribed reagent strips and equipment to self-measure plasma glucose on finger-prick samples, and are instructed on how to use the results both routinely for dose titration and when an unexpected finding occurs[9,10]. More recently, continuous glucose monitoring systems have been approved for use by people on insulin pumps or injections and this technology continues to progress[11–13].
The insulin biobetter cascade
Different stages of the biobetter cascade have had very different impacts in terms of their importance for approval by regulators and adoption by clinicians and patients[2,3].
Better insulin preparations were introduced quite simply by the application of repeated recrystallisation, such that insulin purity went from around 30% in 1922 to around 90% by the end of the 1930s[2]. Such changes might appear to be of little beneficial consequence for clinical practice, but pharmaceutical insulin went from around 10U/mL to 40U/mL and 80U/mL and eventually 100U/mL, with insulin standardisation rising to 24U/mg and later to above 28U/mg.
With the high purity available from the 1970s, manufacturers changed from animal bio-assays to chemical (initially nitrogen) assays and GC-MS, but the standard unit of all preparations remains the same, bar one preparation (see ‘Insulin detemir‘). This might seem odd, as the receptor affinity interaction of the newer designer analogues does vary, but insulin is very rapidly cleared through its receptor, so if affinity and clearance goes down, circulating concentration goes up nearly exactly in proportion, and in vivo potency remains the same[14].
The next step in the cascade was different in nature, although it retained the same insulin (then pork or beef insulin; see Table 1 and Figure 1). While a diverse series of attempts were made to extend the action of a subcutaneous insulin injection, notably in the 1930s. It was a Danish industrial chemist, HC Hagedorn, who suggested that the basic protamine would be a good candidate for holding insulin molecules in a complex, though it took until 1946 to define a stable preparation (NPH) based on the correct molecular ratios and the obligatory presence of phenol/m-cresol[15]. This was both a new chemical entity and not a new chemical entity (all components being familiar) and easily gained regulatory clearance[15].
The Pharmaceutical Journal
Insulin led the way in the manufacturing of biologicals, initially with further purification, this time with the introduction of industrial-scale column chromatography to remove insulin-like and other impurities of pancreatic origin[16]. While this removed the clinical problems of immunological insulin resistance, immunological neonatal hypoglycaemia and lipodystrophy, it became critically important in the early 1980s when insulin production moved to E. coli, and later yeasts, to ensure elimination of cellular components. This was a manufacturing change of a scale that required full regulatory approval, but that matched with the structural change from pork to human sequence insulin, albeit that being only one in 51 amino acid residues. However, the clinical consequences of the manufacturing and structural change were unknown and, in contemporary studies, were of a quality which perhaps would not now be acceptable[17].
Recombinant DNA technology allowed the development of novel insulins, analogues of human insulin[18,19]. Neither human meal-time insulin nor human NPH insulin had appropriate subcutaneous absorption profiles, so modification to alter these characteristics by amino acid substitution was welcome. These were new molecules and missteps occurred in their development, notable with one single amino-acid substituted analogue that was found to promote mammary tumours in rodents[20]. The importance of a full new product assessment when structural change to a biological is made, however minor, should be emphasised[14]. Further developments in the meal-time analogue field have been to formulation alone. Both insulins aspart and lispro now have versions with lower tendency to form insulin hexamers, which are too large to easily enter the vasculature[19,21]. These are formulation changes, and employ excipients to enhance access through the endothelium into the blood. Further formulation changes show promise of even more physiological meal-time profiles, as discussed in the section on use of these insulins below.
However, other structural changes of a more radical nature have produced valuable basal insulin products, notably acylation through a link to the end of the insulin B-chain. These insulins (detemir and degludec) then bind to albumin and delay their absorption from the subcutaneous space and provide a buffer to variable absorption in the form an albumin-bound pool in the circulation[2]. More recently, and with further amino acid changes designed to decrease receptor affinity and thus further increase the circulating reservoir, an insulin developed for weekly administration (insulin icodec) has been in phase III trials[4,22]. Other complexing has been used to extend the action of glucagon-like peptide 1 receptor agonists (GLP-1RA), and notably union with the Fc fragment of immunoglobulin (dulaglutide), which has now been applied to insulin (insulin efsitora alfa) and is also now in phase III trials as a weekly injection candidate[23].
The biobetter cascade can be summarised as steps in purification, manufacturing, structural and formulation change (see Figure 1), but what remains is a therapy that cannot deliver normal blood glucose control in anyone with severely insulin deficient (e.g. type 1) diabetes mellitus.
Current clinical use of insulin preparations – basal insulins
Insulin preparations available in the UK are set out in Table 1, with more detail on basal insulins in Table 2.
NPH insulin
Human NPH insulin is still endorsed by clinical guidelines in the UK for the management of type 2 diabetes mellitus (T2DM) as a starter regimen[10]. Blood glucose control, as measured by glycated haemoglobin (HbA1c), is not improved in insulin starters by insulin glargine 100U/mL or insulin detemir when compared to NPH insulin once daily. Acquisition cost, even compared to biosimilars, is around 30% lower, which might amount to around £10 per month (see Table 2). With increased insulin deficiency, this 12-hour insulin needs to be given twice daily, increasing self-monitoring costs and need for professional interactions. Further, NPH regimens do result in more hypoglycaemia in the same studies used to show no difference in HbA1c, which has increased costs for users (unpleasant, inconvenient, adverse health consequences, such as falls and hypothermia) and thus for the health service[24,25]. However, these factors have little impact on cost effectiveness and because only a small percentage of users experience hypoglycaemia (around 20%), there is always the option to swap to a basal analogue in this group.
More subtly, the increased hypoglycaemia means that the HbA1c measurement with NPH is lowered inappropriately, so overall glucose control may not in fact be equivalent. Funders allow this where twice-daily NPH insulin is needed, but it must be administered by a third party owing to incapacity or disability, then the increased costs of this compared to a once daily analogue are outweighed[10,26].
In practice, if NPH insulin is used as a starter insulin in T2DM, as the condition worsens with time (as is usual), a basal analogue gets substituted before either a GLP-1RA or meal-time insulin is added.
A special example of NPH insulin is found in pre-mixed preparations, that is based on either human insulin or a meal-time analogue; however, the usual cascade of GLP-1RA then basal insulin (perhaps in combination) then addition of meal-time insulin, leaves no space for twice daily pre-mixed regimens whether based on human or analogues insulins. They are however still popular in India and China as starter regimens in people with blood glucose control well above target levels, and then continued indefinitely, despite the problems of increased hypoglycaemia. In the UK, many users persist with them, some clinicians still endorse their use, and they are sometimes used twice daily instead of a meal-time + basal regimen in those who find four injections daily unacceptable[27].
Another important use for NPH insulin is where rapid attainment of glucose control is required in insulin starters, or where insulin requirements may be changing rapidly. This could occur when found in an inpatient where hyperglycaemia is diagnosed before a necessary procedure, where glucose control has been disturbed by concomitant illness, or where metabolic stresses are changing rapidly. Here, the opportunity to adjust and optimise insulin doses twice rather than once daily may be valuable and time-saving, based on bedside finger prick plasma glucose monitoring.
Insulin glargine 100 U/mL and insulin detemir
These human–insulin analogues were developed as once-daily insulins. Insulin glargine, by virtue of addition of basic amino acids, precipitates after injection when neutralised by tissue fluids, forming a dispersed precipitate, which is then slowly absorbed with a half-time of about 12 hours. Insulin detemir was the first albumin-binding acylated insulin to be granted marketing approval. While concern was expressed about the increased IGF-1 receptor activity of glargine, with questions related to malignancy and retinopathy, randomised studies and the observation that the circulating moiety after subcutaneous metabolism has lower rather than high IGF-1 activity have abrogated that problem[4]. The low in-circulation potency of detemir and the resultant four-times higher molecular dosage are not identified with any safety threats[28].
Neither of these insulins is a true 24-hour insulin, which is why they are recommended for evening injection when given once daily, to gain optimal effect at the end of the night. This profile can still be a problem in people with type 1 diabetes mellitus (T1DM) in the late afternoon and evening when basal insulin supply wanes. Solutions are to switch to twice-daily injection (making five injections a day in all), to a true 24-hour preparation or to a pump. This is not such a problem for people with T2DM because their own residual insulin secretion will often buffer shorter periods of insulin insufficiency as glucose concentration rises. Nevertheless, since they were first introduced, these have become the standard basal insulins for people with T1DM, usually in combination with a meal-time insulin analogue. This reflects clinical trials for both showing improved glycaemic control together with less hypoglycaemia compared to a regimen of human NPH and meal-time insulin[29,30]. Use of biosimilar insulin glargine 100U/mL is discussed below.
A problem can arise when swapping away from these insulins when used twice daily; for example, when switching to insulin degludec. In these circumstances, a prospective dose reduction is suggested[31]. A unique issue also arises with insulin detemir, namely that the unit:molar ratio is four times lower than for any other insulin to give equivalent effect; correctly then the detemir unit is designated as such (U) and is not the standard international unit (IU)[32].
Insulin glargine 300 U/mL and insulin degludec
By biobetter category, these competitor insulins are very different. Insulin glargine 300U/mL is a formulation change, the higher concentration giving a denser precipitate after injection and this being absorbed over about 30 hours. However, the circulating entity is the same as for insulin glargine 100U/mL, so the off-target safety data from the very extensive use of that insulin can be applied. Insulin degludec is a novel entity, with a single amino acid substitution and a novel (for insulin) fatty acid linked to the molecule by a spacer[33]. However, in practice both went through full new-entity drug development programmes and review. Degludec is even longer acting than 300U/mL glargine — it is a 24-hour insulin when given once daily[34].
Both of these insulins can be given any time of day, usually and ideally around the same time of day. Morning injection together with the breakfast meal insulin is then possible. With glargine 300U/mL studies show injection time can be varied by up to three hours either way from usual — a situation useful to most people’s lifestyle[35]. With degludec, this interval can be eight hours or even longer[36].
Insulin degludec causes hypoglycaemia in less people with T1DM and T2DM compared to glargine 100U/mL, and indeed less severe hypoglycaemia, a measure for which conventional phase III studies are usually underpowered, but without extra protection against cardiovascular events[37]. Insulin glargine 300U/mL gives less hypoglycaemia in randomised controlled trials in T2DM[38]. However, uptake in T2DM has been relatively muted, though funders agree these insulins can be available to those users with problems when taking the first-generation analogues. Remembering, however, that nearly all people with T1DM have problems with their glucose control when using insulin, degludec has seen more broader use in this population, but clinical experience has not been overwhelmingly of benefit.
Insulin icodec and insulin efsitora alfa — weekly injection candidates
Insulin icodec has already been submitted for regulatory approval, while insulin efsitora alfa (also known as basal insulin Fc) is in phase III studies[22,23]. Phase III company announcements suggest that insulin icodec appears to increase hypoglycaemia, but if it is licensed, the experience with weekly GLP-1 receptor agonists suggests that it will be very popular[39]. Logically it will be easier to persuade people to start a weekly injection than a daily one, and adherence might be improved[22]. The issue with weekly insulins is showing that the pharmacodynamic (PD) profile is such that effect does not vary by the day of the week, but no useful data have been made available for icodec (expected later in 2023), and there are only limited phase II data for insulin-Fc[23,40] .
These insulins would logically be used in T2DM where the injection number is then reduced from 28 per 4 weeks to just 4. In T1DM, this translates to 28 per week reducing to 22 per week, which is clearly not as compelling[22].
Current clinical application of insulin preparations – meal-time insulins
Insulin preparations available in the UK are set out in Table 1, with more detail on meal-time insulins in Table 3.
Human unmodified ‘soluble’ insulin
Human unmodified insulin still has three identifiable roles in diabetes management, 50 years from its introduction. Given intravenously, it has the same pharmacokinetic (PK) and PD properties as the later meal-time analogues, and at lower cost, so can be a choice for addition to intravenous fluids (e.g. for use during surgical procedures or in people unable to eat)[41]. More broadly it is sometimes used in people with T2DM together with NPH insulin as a meal-time + basal insulin regimen, the logic being that this group still has significant endogenous insulin secretion of their own to buffer out the effects of the inappropriate PK profiles[10]. However, by the time people with T2DM come to a multiple injection regimen, they almost by definition have very significant problems with blood glucose control, in which case a longer-acting basal analogue will have been tried, and, by analogy with T1DM if a meal-time insulin is required, a meal-time analogue will be added.
Globally, a huge number of human and animal insulin preparations are approved and regulated, often in local markets. In the UK, porcine, as well as human-sequence insulin, is available, with a handful of users finding they can perceive a difference. Human insulin is also available as a 500U/mL preparation, potentially useful in those needing very high doses, but with a more drawn-out (thus less physiological) absorption profile[42]. These ‘soluble’ insulins have generally been introduced as new entities, before the concept of biosimilars was thought of, though often with brief clinical trial data. That would usually not be unreasonable — the issues would be manufacturing practice and purity, as the PK and safety profiles for a dissolved product would not otherwise be expected to be different. But these insulins are important because production costs are low in some countries, and branded western insulin is expensive when GDP is low. Unfortunately, import, distribution and retail costs often mitigate much of the price advantage in less well-resourced countries.
Meal-time insulin analogues
Insulins lispro and aspart were the original ‘designer’ human-insulin analogues, later joined by insulin glulisine. These are novel molecules, even if only very minor changes to amino acid sequence are made, usually towards the end of the B-chain of the molecule, and not in an area involved in receptor binding[18]. But these developments were not without problems, as one early candidate, which entered studies in humans, was found to cause mammary tumours in rodents, presumably secondary to signalling down the insulin anabolic (growth) pathway[20]. The essence of the ‘designer’ changes is that the insulin hexamer, which cannot be absorbed from the subcutaneous space, is destabilised by dilution after injection, releasing the smaller dimers and monomers (the latter form is receptor active). Accordingly, the PK profile is improved, in the sense of a faster rise after injection, and faster decline as the subcutaneous depot is exhausted, these characteristics being closer to the physiological meal-time plasma concentration profile than is the case for human insulin[25].
Overall, the glucose control profiles are not markedly improved, with small improvements in HbA1c and hypoglycaemia, but in combination with the long-acting analogues, the effects are clinically useful[29,30]. Accordingly, these analogues, together with insulin glargine 100U/mL and insulin detemir, became the standard multiple-injection regimen used in T1DM. This also has become true in T2DM, once transition to a multiple injection regimen is made, as such users will already be taking a long-acting analogue.
Second generation analogue preparations have become available, but these are only formulation changes (see Table 3 and Figure 1)[21]. The issue was to destabilise the hexamer further, without detriment to physico-chemical stability in the insulin vial. There are changes in excipients only, in some cases these also affecting the permeability of the vascular endothelium at the injection site. Since the original molecule is used, terminology can be confusing, as, for example, both this formulation and the original are ‘insulin lispro’. In the medical literature, where proprietary names are frowned upon, a common usage has been ‘faster-acting’ or simply ‘faster’ insulin aspart, for example, though Eli Lilly have encouraged the inappropriate ‘ultra-fast acting insulin lispro’ or ‘URLI’. For dispensing, however, the British National Formulary recommends insulins are prescribed by proprietary name, so the issue does not arise[43].
These second-generation analogue preparations have proved disappointing in practice. Overall glucose control has not been improved and hypoglycaemia is increased in the hour after injection[19]. Post-meal glucose excursions are improved in both T1DM and T2DM, but the clinical significance of this remains uncertain[19]. Because closed-loop systems experience a delay in glucose sensing and a delay in subcutaneous absorption, researchers in this area have been quick to adopt these insulins, but their algorithms are already unstable, resulting in hunting of insulin delivery around a mean.
Development in this area has not stopped, and phase I and II studies of meal-time formulations of the analogues which come even closer to the physiological profile have been reported in the past two years[44]. This now even extends to a 500U/mL preparation, a concentration at which absorption tends to be much slower[45].
Biosimilar insulin use
Background
Biosimilar medicines are not generic medicines. Generics are small, non-biological medicines approved by the regulators if manufacturing processes are assured and the PK properties are unchanged from the parent medicine. Because of the complexity of the molecule, biological medicines are subject to complex manufacturing techniques, and it is difficult to be sure of molecular identity and purity. Accordingly, biosimilar medicines are regulated differently, and the development and proving process is much extended[5,46,47].
Both biosimilar basal and meal-time insulins are available[5]. The use of biosimilar medicines are endorsed and encouraged by funders[46]. Insulin has always been available as ostensibly similar products from different manufacturers from 1923, as was intended by FG Banting, its primary inventor. The licence was made over to Toronto University for US$1, and Eli Lilly’s early contributions to the manufacturing process were also made available through the university[1]. However, it was only with the development of other biologicals, notably erythropoietin and the issue surrounding their copies, that the concept of biosimilars arose and came to be applied to insulin. Unlike insulin copies, biosimilar insulins must follow a detailed regulatory process to show that they have the same cellular properties (pre-clinical), same PK/PD properties (i.e. absorption profile and time effect), and same antigenicity[47]. However, it is assumed that if this is true then all animal and human safety data, and knowledge of clinical application can be carried over from the originator (patented) insulin.
Accordingly, in well-regulated markets, including the UK, biosimilar versions of insulin glargine 100U/mL, insulin lispro and insulin aspart are available, largely owing to the major manufacturers competing among themselves, though one Indian pharmaceutical major (Biocon) has also entered the market. At least one Chinese manufacturer (Gan & Lee) is known to be actively progressing a preparation[5]. Costs of insulin glargine have not fallen significantly (though discounts may be available), but NHS indicative costs of biosimilar lispro and aspart are around 25% lower (see Tables 2 and 3).
Clinical use of biosimilar insulin
NICE generally recommends choosing the insulin with the lowest acquisition cost for any one type, but the situation is complicated by their being three scenarios in which a biosimilar might be substituted for the originator insulin[10,11]. The first is when starting an insulin; there are no issues here, as even if the insulins were subtly different, insulin is individually dose-titrated anyway.
The second is substitution, whereby the diabetes care team suggests change in insulin brand, perhaps on the direction of local funders. It is well recognised that changing brand can be difficult for people with diabetes, who because of erratic glucose control can develop a love-hate relationship with the insulin they are familiar with. Accordingly, this can only be done with increased discussion pre-change, increased self-monitoring, warnings over risk activities, such as driving, and increase contact support after changeover[48]. There is a cost for this, not least for already overstretched services. Further, some insulin users will abreact to the new insulin and must revert to their original insulin type.
The third scenario, which is mandated by law in the United States, but not supported in the UK and Europe, is interchangeability, akin to the pharmacy substitution of generic medicines. The problem here, and why insulin is prescribed by brand not approved name, is the same as guided substitution, namely that specialist support is needed even for any change of brand of the same insulin type[48].
In practice, the biosimilar insulins we have available are manufactured by experienced insulin companies with one exception. Unlike the situation with copies, where in Mexico and India significant problems with biosimilar medications were reported, biosimilar insulins seem to have been well accepted in clinical practice[49,50].
Box: Biosimilar insulins
- Shown to be similar but not identical to originator insulin in pre-clinical chemical and cell studies, phase II PK/PD studies, and phase III clinical (antigenicity) studies;
- Now available for insulins glargine, lispro and aspart;
- Modest, but in some cases useful, cost savings;
- Should be used as starter insulins according to acquisition cost;
- May be substituted in a controlled manner by diabetes teams, but with time and monitoring costs;
- Should be prescribed by brand, and are not interchangeable by pharmacists.
Conclusion
Diverse insulin preparations have been developed over the 100 years of technological advance since the first pancreatic extract, and from the NPH insulin of 1946 (though now in human sequence form), these remain indicated for different groups of people with diabetes to this day. The technological drive does however underline the overall failure of insulin preparations to offer an easy approach to normal circulating glucose concentrations, a problem that continues and is likely to do so. Accordingly, deployment of marketed insulins needs to be tailored to individual need as discussed above, and these needs are not just those of a standard person with T1DM and T2DM. There remains considerable room for improvement both in meal-time insulins (with physiological onset after injection) and basal insulin (peakless weekly profiles), while closed-loop ‘artificial pancreas’ systems struggle to provide feedback on glucose control presently owing to sensing and delivery in a non-physiological (subcutaneous) site.
Biosimilar insulins are welcome for cost containment reasons, and are safe, but substitution must be team-supported, which itself has costs. The cost savings are useful but not substantial, and a problem will be that while technological advance continues, and is needed, insulins out of patent will often not be the optimal choice.
Acknowledgements
This article is unfunded. The author thanks colleagues in clinical practice, academia and industry for fruitful in-depth discussions over four decades, but has not discussed this specific article with any third party.
Duality of interest
The author has been involved in the development of most of the insulins discussed above, and he or institutions with which he is associated have received funding in that regard, and additionally for related research studies and/or lecturing. This includes Biocon, Eli Lilly, Gan & Lee, Novo Nordisk and Sanofi but also manufacturers of competing glucose-lowering products namely AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Janssen, Merck (MSD) and Servier.
- 1Bliss M. The Discovery of Insulin. 1982. doi:10.7208/chicago/9780226075631.001.0001
- 2Home P. The evolution of insulin therapy. Diabetes Research and Clinical Practice. 2021;175:108816. doi:10.1016/j.diabres.2021.108816
- 3Owens DR, Monnier L, Ceriello A, et al. Insulin Centennial: Milestones influencing the development of insulin preparations since 1922. Diabetes Obesity Metabolism. 2021;24:27–42. doi:10.1111/dom.14587
- 4Home PD, Mehta R. Insulin therapy development beyond 100 years. The Lancet Diabetes & Endocrinology. 2021;9:695–707. doi:10.1016/s2213-8587(21)00182-0
- 5Heinemann L, Davies M, Home P, et al. Understanding Biosimilar Insulins – Development, Manufacturing, and Clinical Trials. J Diabetes Sci Technol. 2022;:193229682211058. doi:10.1177/19322968221105864
- 6Goldsmith D, Dellanna F, Schiestl M, et al. Epoetin Biosimilars in the Treatment of Renal Anemia: What Have We Learned from a Decade of European Experience? Clin Drug Investig. 2018;38:481–90. doi:10.1007/s40261-018-0637-1
- 7Dey M, Zhao SS, Moots RJ. Anti-TNF biosimilars in rheumatology: the end of an era? Expert Opinion on Biological Therapy. 2020;21:29–36. doi:10.1080/14712598.2020.1802421
- 8Galloway JA, Spradlin CT, Nelson RL, et al. Factors Influencing the Absorption, Serum Insulin Concentration, and Blood Glucose Responses After Injections of Regular Insulin and Various Insulin Mixtures. Diabetes Care. 1981;4:366–76. doi:10.2337/diacare.4.3.366
- 9Type 1 diabetes in adults: diagnosis and management. National Institute for Health and Care Excellence. 2015.www.nice.org.uk/guidance/ng17 (accessed Jul 2023).
- 10Type 2 diabetes in adults: management. National Institute for Health and Care Excellence. 2015.www.nice.org.uk/guidance/ng28 (accessed Jul 2023).
- 11Diabetes (type 1 and type 2) in children and young people: diagnosis and management. National Institute for Health and Care Excellence. 2015.www.nice.org.uk/guidance/ng18 (accessed Jul 2023).
- 12Continuous subcutaneous insulin infusion for the treatment of diabetes mellitus. Technology appraisal guidance [TA151]. National Institute for Health and Care Excellence. 2008.www.nice.org.uk/guidance/ta151 (accessed Jul 2023).
- 13Hybrid closed loop systems for managing blood glucose levels in type 1 diabetes. Appraisal consultation document. National Institute for Health and Care Excellence. 2023.www.nice.org.uk/guidance/indevelopment/gid-ta10845 (accessed Jul 2023).
- 14Kurtzhals P, Schäffer L, Sørensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes. 2000;49:999–1005. doi:10.2337/diabetes.49.6.999
- 15Gabriele AJ, Marble A. Clinical experience with a new modified protamine insulin (NPH-50). Am. J. Dig. Dis. 1949;16:197–206. doi:10.1007/bf03005026
- 16Schlichtkrull J, Brange J, Hein Christiansen A, et al. Clinical Aspects of Insulin–Antigenicity. Diabetes. 1972;21:649–56. doi:10.2337/diab.21.2.s649
- 17Richter B, Neises G. ‘Human’ insulin versus animal insulin in people with diabetes mellitus. Cochrane Database of Systematic Reviews. 2005;2010. doi:10.1002/14651858.cd003816.pub2
- 18Brange J, Ribel U, Hansen JF, et al. Monomeric insulins obtained by protein engineering and their medical implications. Nature. 1988;333:679–82. doi:10.1038/333679a0
- 19Bolli GB, Porcellati F, Lucidi P, et al. One-hundred year evolution of prandial insulin preparations: From animal pancreas extracts to rapid-acting analogs. Metabolism. 2022;126:154935. doi:10.1016/j.metabol.2021.154935
- 20Falck HANSEN B, DANIELSEN GM, DREJER K, et al. Sustained signalling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochemical Journal. 1996;315:271–9. doi:10.1042/bj3150271
- 21Avgerinos I, Papanastasiou G, Karagiannis T, et al. Ultra‐rapid‐acting insulins for adults with diabetes: A systematic review and meta‐analysis. Diabetes Obesity Metabolism. 2021;23:2395–401. doi:10.1111/dom.14461
- 22Rosenstock J, Del Prato S. Basal weekly insulins: the way of the future! Metabolism. 2022;126:154924. doi:10.1016/j.metabol.2021.154924
- 23Frias J, Chien J, Zhang Q, et al. Safety and efficacy of once-weekly basal insulin Fc in people with type 2 diabetes previously treated with basal insulin: a multicentre, open-label, randomised, phase 2 study. The Lancet Diabetes & Endocrinology. 2023;11:158–68. doi:10.1016/s2213-8587(22)00388-6
- 24Riddle MC, Rosenstock J, Gerich J, et al. The Treat-to-Target Trial. Diabetes Care. 2003;26:3080–6. doi:10.2337/diacare.26.11.3080
- 25Hermansen K, Davies M, Derezinski T, et al. A 26-Week, Randomized, Parallel, Treat-to-Target Trial Comparing Insulin Detemir With NPH Insulin as Add-On Therapy to Oral Glucose-Lowering Drugs in Insulin-Naïve People With Type 2 Diabetes. Diabetes Care. 2006;29:1269–74. doi:10.2337/dc05-1365
- 26Medicines advice: insulin glargine, SMC11/02. Scottish Medicines Consortium. 2002.www.scottishmedicines.org.uk/medicines-advice/ (accessed Jul 2023).
- 27Twice-daily insulin regimen. Diabetes UK. 2019.www.diabetes.co.uk/insulin/twice-daily-insulin-regimen.html (accessed Jul 2023).
- 28Home P, Kurtzhals P. Insulin detemir: from concept to clinical experience. Expert Opinion on Pharmacotherapy. 2006;7:325–43. doi:10.1517/14656566.7.3.325
- 29Hermansen K, Fontaine P, Kukolja KK, et al. Insulin analogues (insulin detemir and insulin aspart) versus traditional human insulins (NPH insulin and regular human insulin) in basal-bolus therapy for patients with Type 1 diabetes. Diabetologia. 2004;47:622–9. doi:10.1007/s00125-004-1365-z
- 30Ashwell SG, Amiel SA, Bilous RW, et al. Improved glycaemic control with insulin glargine plus insulin lispro: a multicentre, randomized, cross-over trial in people with Type 1 diabetes. Diabet Med. 2006;23:285–92. doi:10.1111/j.1464-5491.2005.01781.x
- 31Summary of Product Characteristics, Tresiba (revised). Novo Nordisk. Electronic Medicines Compendium. 2022.www.medicines.org.uk/emc/product/7936/smpc (accessed Jul 2023).
- 32Levemir EPAR (renewal). European Medicines Agency. 2009.www.ema.europa.eu/en/documents/product-information/levemir-epar-product-information_en.pdf (accessed Jul 2023).
- 33Jonassen I, Havelund S, Hoeg-Jensen T, et al. Design of the Novel Protraction Mechanism of Insulin Degludec, an Ultra-long-Acting Basal Insulin. Pharm Res. 2012;29:2104–14. doi:10.1007/s11095-012-0739-z
- 34Heise T, Hermanski L, Nosek L, et al. Insulin degludec: four times lower pharmacodynamic variability than insulin glargine under steady‐state conditions in type 1 diabetes. Diabetes Obes Metab. 2012;14:859–64. doi:10.1111/j.1463-1326.2012.01627.x
- 35Riddle MC, Bolli GB, Home PD, et al. Efficacy and Safety of Flexible Versus Fixed Dosing Intervals of Insulin Glargine 300 U/mL in People with Type 2 Diabetes. Diabetes Technology & Therapeutics. 2016;18:252–7. doi:10.1089/dia.2015.0290
- 36Mathieu C, Hollander P, Miranda-Palma B, et al. Efficacy and Safety of Insulin Degludec in a Flexible Dosing Regimen vs Insulin Glargine in Patients With Type 1 Diabetes (BEGIN: Flex T1): A 26-Week Randomized, Treat-to-Target Trial With a 26-Week Extension. The Journal of Clinical Endocrinology & Metabolism. 2013;98:1154–62. doi:10.1210/jc.2012-3249
- 37Marso SP, McGuire DK, Zinman B, et al. Efficacy and Safety of Degludec versus Glargine in Type 2 Diabetes. N Engl J Med. 2017;377:723–32. doi:10.1056/nejmoa1615692
- 38Ritzel R, Roussel R, Giaccari A, et al. Better glycaemic control and less hypoglycaemia with insulin glargine 300 U/mL vs glargine 100 U/mL: 1-year patient-level meta-analysis of the EDITION clinical studies in people with type 2 diabetes. Diabetes Obes Metab. 2017;20:541–8. doi:10.1111/dom.13105
- 39Insulin icodec press releases. Novo Nordisk. https://www.novonordisk.com/news-and-media/news-and-ir-materials.html (accessed Jul 2023).
- 40Home P. Making sense of weekly insulins. The Lancet Diabetes & Endocrinology. 2023;11:140–1. doi:10.1016/s2213-8587(23)00002-5
- 41Human Insulin. Diabetes UK. 2019.https://www.diabetes.co.uk/insulin/human-insulin.html (accessed Jul 2023).
- 42Sze D, Goldman J. Human Regular 500 units/mL Insulin Therapy: A Review of Clinical Evidence and New Delivery Options. Clinical Diabetes. 2018;36:319–24. doi:10.2337/cd18-0004
- 43Insulin. British National Formulary. https://bnf.nice.org.uk/drugs/insulin/ (accessed Jul 2023).
- 44Svehlikova E, Mursic I, Augustin T, et al. Pharmacokinetics and Pharmacodynamics of Three Different Formulations of Insulin Aspart: A Randomized, Double-Blind, Crossover Study in Men With Type 1 Diabetes. Diabetes Care. 2020;44:448–55. doi:10.2337/dc20-1017
- 45Svehlikova E, Ashcroft NL, Gatschelhofer C, et al. Pharmacokinetics and Pharmacodynamics of a Novel U500 Insulin Aspart Formulation: A Randomized, Double-Blind, Crossover Study in People With Type 1 Diabetes. Diabetes Care. 2023;46:757–64. doi:10.2337/dc22-1054
- 46What is a biosimilar medicine? . NHS England. 2023.www.england.nhs.uk/long-read/what-is-a-biosimilar-medicine/ (accessed Jul 2023).
- 47Non-clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues – scientific guideline. European Medicines Agency. 2015.www.ema.europa.eu/en/non-clinical-clinical-development-similar-biological-medicinal-products-containing-recombinant-human (accessed Jul 2023).
- 48Biosimilar insulins: position statement. Diabetes UK. 2019.www.diabetes.org.uk/professionals/position-statements-reports/diagnosis-ongoing-management-monitoring/biosimilar-insulins (accessed Jul 2023).
- 49Owens DR, Landgraf W, Schmidt A, et al. The Emergence of Biosimilar Insulin Preparations—A Cause for Concern? Diabetes Technology & Therapeutics. 2012;14:989–96. doi:10.1089/dia.2012.0105
- 50Malhotra H. Biosimilars and non-innovator biotherapeutics in India: An overview of the current situation. Biologicals. 2011;39:321–4. doi:10.1016/j.biologicals.2011.06.018