Science Photo Library
A tiny globule of fat suspended in a fine mist makes its way along the branching passageways of a human lung. Deep in the lung the globule, called a liposome, nestles up against one of the cells lining the airway. Because the cell membrane is also made of fat, the liposome is able to pass through it, and spill its cargo into the interior of the cell: a string of DNA encoding a protein known as CFTR, which will enable the cell to make thin, watery mucus that helps keep the lung clean and healthy.
This is gene therapy for cystic fibrosis, a genetic disorder that causes serious lung disease. In a phase III trial published in 2015, the UK Cystic Fibrosis Gene Therapy Consortium, a group of scientists and clinicians from London, Oxford, and Edinburgh, reported that inhaling these liposomes every month for a year stabilised the lung function of individuals with cystic fibrosis[1]
. Meanwhile, lung function deteriorated slightly in the study’s placebo group.
These are modest effects, as even the study’s authors acknowledge. “I think we need to improve on that” before the therapy could be rolled out to clinical care, says Eric Alton, professor of gene therapy and respiratory medicine at Imperial College London and the consortium’s coordinator.

Source: Eric Alton
Eric Alton, professor of gene therapy and respiratory medicine at Imperial College London, is coordinator of the UK Cystic Fibrosis Gene Therapy Consortium
Still, the trial represented a huge milestone for the field. For the first time, researchers had demonstrated the efficacy of cystic fibrosis gene therapy in humans — a goal that has seemed out of reach for more than a quarter of a century.
The success reflects not only the pay-off of persistence but the current buzz of activity in the field. A range of approaches are in play, including viral vectors as well as non-viral ones like liposomes for delivery, and classic gene therapy strategies that would add healthy genetic material to a patient’s cells, as well as cutting-edge technologies that would edit their own genetic material. So far, most of the current crop of potential approaches have only been tested in cells grown in laboratory dishes or in animal models. But all this activity reflects the field’s renewed optimism that somehow, correcting the genetic defect that causes cystic fibrosis will soon be possible.
Prototype disease
Cystic fibrosis is caused by mutations in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR). This protein sits in the cell membrane and helps regulate the flux of water and negatively charged chloride ions into and out of cells.
CFTR’s broad function means that the disease affects a variety of organs, including the lung, pancreas, liver, intestine and testes. Without functional CFTR protein, the body has trouble making sweat, digestive fluids and mucus normally. However, most people with the disease die from lung complications: the mucus that clogs the airways of cystic fibrosis patients makes their lungs vulnerable to infection and scarring, and even in developed countries half of patients die before the age of 40 years.
Cystic fibrosis was really at that moment the prototype disease to be used for gene therapy
The CFTR gene was identified in 1989, touching off a wave of enthusiasm for developing a gene therapy cure for the disorder. “Cystic fibrosis was really at that moment the prototype disease to be used for gene therapy,” says Marianne Carlon, a gene therapy researcher at Catholic University in Leuven, Belgium.
“Theoretically, cystic fibrosis is easy,” says Carlon. It’s a recessive, single-gene disorder, meaning that two mutated copies of CFTR are necessary to produce the disease — and, by extension, delivering a single functional copy of that gene to airway cells ought to alleviate it. And in the lung, gene therapy can be delivered by inhalation rather than more invasive methods.

Source: Marianne Carlon
Marianne Carlon, a gene therapy researcher at Catholic University in Leuven, Belgium, says liposomes are not very efficient at getting their genetic cargo into the host nucleus
Cystic fibrosis was also a compelling target because it is one of the most common lethal inherited diseases, with more than 90,000 sufferers worldwide, so gene therapy could help many people. Between 1993 and 2015, scientists conducted more than two dozen clinical trials involving about 450 patients[2]
. Most of these were single-dose studies, and were designed to assess safety or molecular function, not clinical benefit.
The very fact that the lung is accessible through the airways to the environment means that it has a vast array of defense mechanisms
But researchers quickly discovered that gene therapy for cystic fibrosis is more complicated in practice. “The very fact that the lung is accessible through the airways to the environment means that it has a vast array of defense mechanisms,” says Chris Boyd, a medical geneticist at the University of Edinburgh and a member of the UK consortium. These defense mechanisms mean that the cells lining the airways — exactly the cells most in need of a functional copy of CFTR— perceive gene therapy delivery systems as alien invaders, and do their best to keep them out.

Source: Chris Boyd
Chris Boyd, a medical geneticist at the University of Edinburgh and a member of the UK Cystic Fibrosis Gene Therapy Consortium, says the cells lining the airways perceive gene therapy delivery systems as alien invaders, and do their best to keep them out
The delivery problem is even worse in the lungs of cystic fibrosis patients because the thick, sticky mucus characteristic of the disease presents an additional barrier to airway cell entry.
More challenging still, gene therapy for cystic fibrosis has to be performed repeatedly throughout a person’s life, since airway cells bearing the functional gene die off and are replaced by new, uncorrected ones. And the immune system learns to recognise and fight off the viruses that are often used as gene therapy vectors, so the more a treatment is administered, the less effective it becomes.
“In reality, cystic fibrosis proved to be one of the most difficult targets you could ever imagine,” says Alton.
These frustrations caused many of the initial wave of researchers to abandon the field. In the past few years, however, improvements in gene therapy tools and technologies, as well as some encouraging successes in developing gene therapies for other diseases, have sparked a resurgence of interest in cystic fibrosis gene therapy.
CFTR correctors
Meanwhile, the landscape of cystic fibrosis treatment changed radically with the development of CFTR correctors, which interact with mutant CFTR proteins to improve their function[3]
. “They’re fabulous products, and it’s really quite a revolution in cystic fibrosis care,” says David Parsons, who heads the cystic fibrosis gene therapy team at the University of Adelaide in Australia. These drugs have raised the bar for gene therapy because they target the underlying molecular defect, rather than merely treating cystic fibrosis symptoms as past treatments have done.
But the drugs only help patients with a few of the nearly 2,000 known CFTR mutations. “That leaves out a lot of people,” says Parsons.
The most impressive CFTR corrector so far, ivacaftor, is most effective in those with so-called class III mutations, who total around 4% of cystic fibrosis patients. A second formulation — a combination of ivacaftor and another molecule, lumacaftor — targets the most common mutation, known as Phe508del, which accounts for about 70% of CFTR mutations in Caucasians. But its effectiveness is much more modest.
This leaves a big opening for gene therapy, since the majority of approaches to that treatment would help regardless of the mutation a patient carries.
The UK consortium’s trial, published in 2015, was open to patients with any CFTR mutation, for example (although the majority carried Phe508del, as expected given its prevalence)[1]
. The team chose a liposome vector because, unlike viruses, liposomes do not trigger an immune reaction and so can be administered over and over again.
But the approach has its disadvantages. “Liposomes are nice for a proof of concept,” says Carlon, who was not involved in the work. However, they “really are not very efficient at getting their genetic cargo into the host nucleus”.
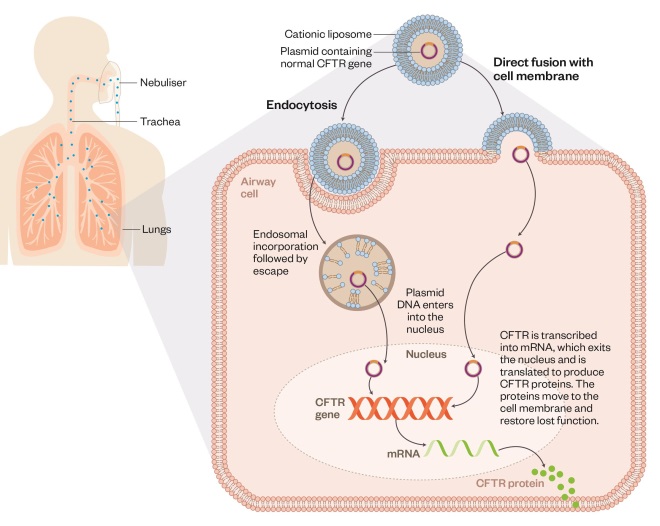
Using liposomes to deliver gene therapy in cystic fibrosis
Editorial adviser: Chris Boyd
Patients with cystic fibrosis lack functional CFTR protein, which is needed to make thin, watery mucus that keeps the lungs healthy. Normal CFTR gene can be delivered via inhalation of nebulised liposomes mixed with plasmid DNA encoding the protein. The liposomes enhance transport of the CFTR gene into host airway cells via endocytosis or direct fusion with the cell membrane. The plasmid then passes into the cell nucleus where the normal CFTR is transcribed, enabling the cell to make CFTR proteins.
The UK consortium researchers suspect that this lack of efficiency accounts for the modest clinical results seen in the trial. They say the effectiveness may be improved by delivering a larger dose to the lungs with each gene therapy treatment.
Several other groups around the world are working on other non-viral approaches to gene therapy, such as DNA nanoparticles, in which genetic material is tightly packed within a polymer coating. These nanoparticles may be especially good at penetrating the thick mucus layer in the lungs of cystic fibrosis patients[4]
.
Lentiviral vectors
At the same time, recent advances in viral vectors for gene therapy are causing the cystic fibrosis field to take another look at this strategy. “A virus knows how to get DNA to the nucleus — because of course that’s what viruses do, that’s how they’ve been programmed by evolution,” says Alton. “And so why would you not use a viral vector if you could?”
A virus knows how to get DNA to the nucleus — because of course that’s what viruses do, that’s how they’ve been programmed by evolution
Lentiviruses have shown promise in delivering gene therapy for other diseases, such as inherited immune deficiencies. For reasons scientists do not understand, viruses in this class do not seem to trigger the type of immune response that prevents gene therapy from being administered multiple times, so they could be a good match for cystic fibrosis as well. Multiple research groups worldwide are developing gene therapy for cystic fibrosis using lentiviral vectors.
The UK consortium’s entry in this field is based on the simian immunodeficiency virus (SIV). In collaboration with a Japanese biotechnology company, they have engineered a SIV-based vector with two proteins from a mouse respiratory virus, Sendai virus, which makes it extremely efficient at getting into airway cells[5]
.
Lentiviral vectors may also yield longer lasting gene correction because the genetic material they deliver becomes permanently integrated into the host cell’s genome. However, this advantage also comes with a safety concern, as the genetic material could splice in at a harmful location and cause cancer. Lentiviral vectors are thought to be safer in this regard than viruses used in the early days of gene therapy, although lentiviruses have never been delivered to the human airway. The UK consortium plans to begin a trial on this in late 2017.
The outcome of that trial will determine the fate of the group’s liposome work: if the greater efficiency of the lentiviral vector pans out, the consortium will focus its efforts there, but this isn’t yet certain.
Parsons says the consortium’s viral-vector results so far look “very promising”. For the past decade, he has been working on a different lentiviral vector for cystic fibrosis gene therapy, based on the human immunodeficiency virus (HIV). Parsons’s protocol involves treating the airways just prior to gene therapy with lysophosphatidylcholine (LPC), a molecule that causes the tight junctions between airway cells to open briefly[6]
.
This opening enables the gene therapy vector to reach the underside of airway cells, where this particular vector happens to be able to enter more easily. It also allows access to the stem cells that renew the airway lining, which are typically located beneath the surface of the airway.
“If you can target stem cells it changes the game,” says Parsons. Delivering a healthy copy of CFTR to airway stem cells could permit one-time gene therapy leading to permanent correction of the defect.
LPC is a molecule normally found in the lungs. However, opening the tight junctions between airway cells, even briefly, could be risky — especially in the already compromised and often bacteria-infected lungs of cystic fibrosis patients. Parsons says the procedure doesn’t cause harm in animal studies, and has a five-year plan to begin human trials.
Adeno-associated viral vectors
Other researchers are taking a new look at adeno-associated virus (AAV), a vector that was used in some early cystic fibrosis gene therapy trials but did not show effectiveness. More recently, “AAV gene therapy has been shown to be successful in other disease areas,” says Carlon, who is working on one AAV project at Catholic University. “I think that really fuelled reinvestigating this vector system for cystic fibrosis,” she adds.
AAV gene therapy has been shown to be successful in other disease areas — I think that really fuelled reinvestigating this vector system for cystic fibrosis
Early work with AAV used a form of the virus called AAV2, which isn’t very efficient at getting into airway cells. The Leuven team is working with an improved form called AAV5. They also added a stronger promoter, a region of DNA that enables a gene to be turned on.
Animal studies suggest that the vector can produce long-lasting gene expression — at least 15 months in the mouse[7]
. And the researchers say they do not see a fall off in gene expression caused by an immune response to the vector, even after three doses of the therapy[8]
. Next, the team plans to test whether the AAV therapy can improve lung function in a pig or ferret model of cystic fibrosis.
Additional strategies for correcting the CFTR gene make use of the latest molecular techniques. Researchers at Johns Hopkins University in Baltimore, Maryland, have packaged a truncated version of CFTR in an AAV vector that works by transcomplementation[9]
. In this process, small fragments of healthy CFTR protein encoded by the genetic material in the vector interact with the host cell’s mutant CFTR and improve its function.
In essence, this strategy gives cells the ability to make their own CFTR corrector drugs. And, as with CFTR correctors, each transcomplementation-based gene therapy product will only target one or a subset of CFTR mutations. “It’s working very well on Phe508del,” reports Liudmila Cebotaru, who is leading the project at Johns Hopkins University. But some mutations may not be able to be rescued by this strategy.

Source: Liudmila Cebotaru
Liudmila Cebotaru, a researcher at Johns Hopkins University, has packaged a truncated version of CFTR in an adeno-associated virus vector that works by transcomplementation
Other approaches using gene-editing technologies, such as CRISPR, TALENs, and zinc-finger nucleases, are also mutation-specific[10]
. But gene editing, which fixes a patient’s own DNA, has some advantages over gene addition, the usual strategy for gene therapy that simply adds a healthy copy of a gene to a patient’s genome. “You’ll get the subtleties of the gene back as well,” such as when and in what amount and in what form the gene is expressed, says Patrick Harrison, who is working on this approach at University College Cork in Ireland.
Harrison’s current work targets three CFTR mutations that can all be ameliorated by cutting a few DNA base pairs out of the mutated gene. The counterintuitive strategy is a good test case for gene editing because it only requires a single step, cutting the DNA, rather than also inserting a corrected sequence. “It will be more efficient and it will be simpler,” Harrison says. “It will also work in cells that aren’t dividing very quickly, which is often the case in lung tissue.”
The only gene-editing strategy for cystic fibrosis that has been tested in humans so far is QR-010, a molecule developed by the Dutch biotechnology company ProQR to correct the Phe508del mutation, which has just entered phase I clinical trials. The company’s approach is unique in that its gene therapy aims to edit not the mutant DNA itself but the messenger RNA that the cell produces as a template for building a protein[3]
.
On account of its complexity, gene editing will be difficult to take from research tool to clinic, but that doesn’t mean it and any other avenues shouldn’t be investigated. “I think it’s very good for the field that there are lots of different approaches,” says Alton. “It doesn’t matter which of us gets there, it’s just important for the patients that one of us gets there.”
| Drug class | Mechanism | Examples |
|---|---|---|
| Bronchodilators | Widen the airways to help other medications reach the lungs. | Salbutamol, salmeterol. |
| Mucus thinners | Make mucus less sticky so it is easier to remove from lungs through chest physical therapy or coughing. | Hypertonic saline, dornase alpha, mannitol. |
| Pancreatic enzymes | Help the body to digest food and absorb nutrients (replacing enzymes normally produced by the body that cannot always reach the digestive system because they are blocked by thick, sticky mucus). | Pancreatin. |
| Antibiotics | Fight bacterial infections that cystic fibrosis patients are prone to because the thick mucus in their airways tends to trap bacteria. | Many classes of antibiotics are used in cystic fibrosis patients. Patients often take oral or inhaled antibiotics daily to prevent infection, and may need intravenous antibiotics if an infection does take hold. |
| CTFR correctors | For patients with class III mutations in the gene that encodes cystic fibrosis transmembrane conductance regulator (CFTR), accounting for about 4% of cystic fibrosis cases: the drug helps the CFTR protein carry chloride ions across the cell membrane. | Ivacaftor (Kalydeco). |
| CTFR correctors | For patients with two copies of the most common cystic fibrosis mutation, Phe508del: the drug helps shepherd the CFTR protein to the surface of the cell, and ivacaftor improves its chloride transport ability. | Lumacaftor/ivacaftor (Orkambi). |
References
[1] Alton EW, Armstrong DK, Ashby D et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med 2015;3:684–691. doi: 10.1016/S2213-2600(15)00245-3
[2] Griesenbach U, Pytel KM & Alton EW. Cystic fibrosis gene therapy in the UK and elsewhere. Human Gene Therapy 2015;26:266–275. doi: 10.1089/hum.2015.027
[3] Dhooge B, Haaf JB, Noel S et al. Strategies in early clinical development for the treatment of basic defects of cystic fibrosis. Expert Opin Investig Drugs 2016;25:423–436. doi: 10.1517/13543784.2016.1154041
[4] Suk JS, Kim AJ, Trehan K et al. Lung gene therapy with highly compacted DNA nanoparticles that overcome the mucus barrier. J Control Release 2014;178:8–17. doi: 10.1016/j.jconrel.2014.01.007
[5] Griesenbach U, Inoue M, Meng C et al. Assessment of F/HN-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. Am J Respir Crit Care Med 2012;186:846–856. doi: 10.1164/rccm.201206-1056OC
[6] Cmielewski P, Farrow N, Donnelley M et al. Transduction of ferret airway epithelia using a pre-treatment and lentiviral gene vector. BMC Pulm Med 2014;14:183. doi: 10.1186/1471-2466-14-183
[7] Vidović D, Gijsbers R, Quiles-Jimenez A et al. Noninvasive imaging reveals stable transgene expression in mouse airways after delivery of a nonintegrating recombinant adeno-associated viral vector. Hum Gene Ther 2016;27:60–71. doi: 10.1089/hum.2015.109
[8] Carlon MS, Vidović D, Dooley J et al. Immunological ignorance allows long-term gene expression after perinatal recombinant adeno-associated virus-mediated gene transfer to murine airways. Hum Gene Ther 2014;25:517–528. doi: 10.1089/hum.2013.196
[9] Cebotaru L & Guggino WB. Complement yourself: Transcomplementation rescues partially folded mutant proteins. Biophys Rev 2014;6:169–180. doi: 10.1007/s12551-014-0137-3
[10] Lee CM, Flynn R, Hollywood JA et al. Correction of the ΔF508 mutation in the cystic fibrosis transmembrane conductance regulator gene by zinc-finger nuclease homology-directed repair. Biores Open Access 2012;1:99–108. doi: 10.1089/biores.2012.0218