Mclean
Instead of trying to mop up disease-causing proteins once they have been produced, like most small molecule or antibody therapies, gene-silencing treatments target messenger RNA (mRNA) to cut off production of these proteins at source. And pioneering pharmaceutical companies working on this technology now have neurodegenerative diseases in their sights.
Neurodegenerative diseases — such as Alzheimer’s, Parkinson’s and Huntington’s — are common and most lack disease-modifying therapies or cures.
At least 1 in 14 of the UK population aged over 65 years has dementia, with 60–70% of those cases classed as Alzheimer’s disease[1,2]. Data also show that 1 in 37 people in the UK will be diagnosed with Parkinson’s disease in their lifetime, and 1 in 10,000 will develop Huntington’s disease[3,4].
In December 2021, Ionis Pharmaceuticals, based in Carlsbad, California, in partnership with Biogen, based in Cambridge, Massachusetts, published data from the first trial of a gene-silencing drug — BIIB080 — for mild Alzheimer’s disease, showing it to reduce key Alzheimer’s biomarkers and cause no serious adverse events[5].
However, the publication of the Ionis trial came on the heels of two high-profile gene-silencing drug failures for Huntington’s disease, leaving those affected by the condition despondent. Understanding these failures may go some way to improving the next generation of gene-silencing drugs.
“There are molecules that are in the wings, that are longer lasting, safer and more potent,” says chemist Jonathan Watts, associate professor at the RNA Therapeutics Institute at the University of Massachusetts Medical School.
How do they work?
Gene-silencing therapies act to stop the translation of mRNA, providing a method to remove the harmful proteins formed from mutated genes that can lead to disease. The most established method uses antisense oligonucleotides (ASO) — short synthetic single strands of DNA and RNA able to knock down genetic expression through base-pairing with their corresponding ‘sense’ strands of mRNA, before they can be translated into proteins (see Figure).
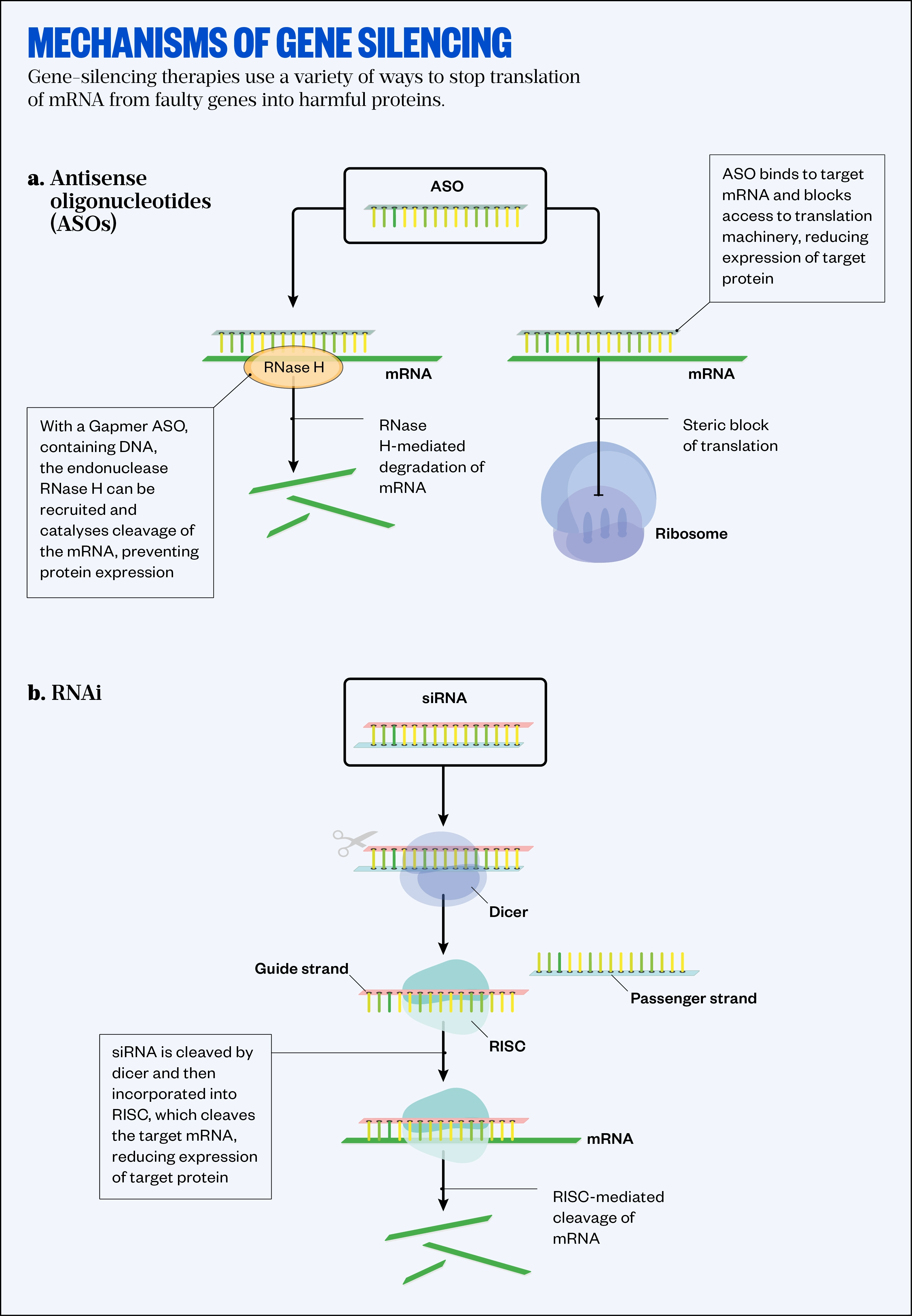
RNase H: Ribonuclease H; RNAi: RNA interference; siRNA: small interfering RNA; RISC: RNA-induced silencing complex
Since 2017, ASOs to treat spinal muscular atrophy (nusinersen [Spinraza; Ionis Pharmaceuticals]) and Duchenne’s muscular dystrophy (casimersen [Amondys 45; Sarepta Therapeutics]) have led the way. Part of the attraction is the simplicity of designing high affinity drugs this way.
“Everybody can match As with Us and Cs with Ts,” jokes Watts.
ASOs can simply block access to the RNA base sequence to stop translation, but a new generation of ASOs, known as Gapmers, are exploiting another mechanism. They are made up of short DNA sequences with RNA-like segments on each side to provide higher affinity to the target, plus resistance to degradation by nucleases. When they bind to mRNA transcripts they are able to recruit an enzyme called Ribonuclease H, which will trigger the RNA’s destruction (see Figure).
Ionis Pharmaceuticals Alzheimer’s ASO BIIB080 was designed to lower levels of tau protein, which when misfolded is thought to cause the neurofibrillary plaques found in patients’ brains. In the phase I trial, launched in 2017, it was administered intrathecally to 44 patients aged 50–74 years with mild Alzheimer’s disease over three months and showed an ability to reduce tau and associated protein markers in the cerebrospinal fluid by 30–50%[5].
The impact on cognition will not be clear until results of the phase II trial, which is set to start in summer 2022, with an estimated completion date of December 2026[6].
Ionis is also developing an ASO drug for Parkinson’s disease — ION464 — designed to prevent the production of the alpha-synuclein protein, known to accumulate in the brains of people with the condition, and has licensed another — tofersen, for a rare, genetic form of amyotrophic lateral sclerosis (ALS) called superoxide dismutase 1 (SOD1) ALS — to Biogen.
In July 2022, the US Food and Drug Administration (FDA) approved a new drug application and accelerated approval pathway for tofersen, despite the drug failing to meet the primary end point in a phase III trial in 2021[7]. Integrated 12-month data from two trials included in the FDA filing suggest that individuals who start tofersen earlier experience a slower rate of decline.
Past failures
The promise of this gene-silencing approach has been tempered by the failures of two ASO drugs to treat Huntington’s disease — a rare genetic condition that causes the progressive degeneration of nerve cells in the brain.
One drug, manufactured by Massachusetts-based Wave Life Sciences, was designed to target the mutant Huntington’s gene (HTT) by recognising a single nucleotide polymorphism (an approach that only works in a subset of patients). However, in an early phase Ib/IIa trial, the drug failed to significantly lower levels of the mutant protein in the cerebrospinal fluid[8].
Roche’s ASO tominersen (licensed from Ionis Pharmaceuticals in 2017) was designed to reduce both the mutant and healthy versions of the protein, but even though a previous phase I/II trial did show lower levels of the protein in treated patients, it failed in its phase III trial of 800 patients in 2021, producing worse outcomes, with five cases of hydrocephalus — a build-up of fluid on the brain — in patients receiving the highest dosages of 120mg[9,10].
Does this indicate a fundamental failure with the approach? “The answers are a definitive no,” says Edward Wild, professor of neurology and consultant neurologist at the National Hospital for Neurology and Neurosurgery, Queen Square, and associate director of University College London’s Huntington’s Disease Centre, who was involved in the tominersen trial.
What both failures show in different ways is how difficult it is to successfully deliver ASOs to the brain. Wild says Wave Life Sciences’ drug failed to engage the target. “That’s really a question of going back to the chemistry of the ASOs and making them better.”
Tominersen, however, did reach its target, but the high doses given to facilitate delivery deep into the basal ganglia caused toxicity, triggering an inflammatory response in the brain.
I think the doses that were chosen were too high; in retrospect, we would do it differently
Edward Wild, a consultant neurologist and associate director of University College London’s Huntington’s Disease Centre
“I think the doses that were chosen were too high,” says Wild. “In retrospect, we would do it differently.”
In subsequent analysis, Roche found that those patients at an earlier stage of the disease, or who were younger, did better than the group as a whole, suggesting they were more able to tolerate the high ASO doses and gain a benefit[11]. As a result, Roche plans to repeat a dose-ranging trial in this patient sub-group to try and test that hypothesis.
The failed drugs were based on chemistry developed in the 1980s and, according to Matthew Wood, professor of neuroscience in the Department of Paediatrics at the University of Oxford, “there are many new chemical improvements that have come along in the past decade”.
Delivery dilemmas
The brain is protected by the blood–brain barrier that prevents entry to most molecules. For this reason, current ASOs are injected directly into the spinal fluid, which provides better access to the brain but makes frequent administration difficult.
A second generation of ASOs include chemical modifications, with the non-bridging oxygens of the oligonucleotide phosphate backbone replaced with sulphur atoms (known as phosphorothioate modification). This improves their ability to resist attack by nucleases and also enhances protein binding and cell uptake properties, as well as overall bioavailability. So, although these drugs will still need to be administered intrathecally, a lower dose is likely to be effective, providing an improved therapeutic window.
The other approach to gene silencing takes advantage of RNA interference, one of the cells’ natural defenses against viral RNA to prevent it being translated into proteins. It works using a multi-protein assembly called RNA-induced silencing complex (RISC), which is able to break down double-stranded RNA to prevent translation. The complex loads one of the strands of an RNA molecule (the anti-sense strand) and here it acts as a template. Any complementary RNA transcripts in the cell will be destroyed before a protein can be formed from them (see Figure).
This can be exploited therapeutically if small interfering RNA (siRNA) molecules are delivered to cells. Here, they will automatically be loaded into RISC and the anti-sense strand will stay there, bound for months, continuing to remove any complementary mRNA produced by the cell. This means continued dosing is not needed but getting the siRNA into the cell in the first place is even more difficult than for an ASO, owing to its larger size and its negatively charged surface.
“If you inject a single stranded ASO into the bloodstream, it is taken up pretty effectively … but a duplex RNA, something that’s fully double stranded, does not show much cell uptake,” explains Watts.
siRNAs are probably of the order of somewhere between one and ten-fold more potent than an ASO system, in theory
Matthew Wood, professor of neuroscience in the Department of Paediatrics at the University of Oxford
siRNA’s ability to silence a gene is much greater though, “probably of the order of somewhere between one and ten-fold more potent than an ASO system, in theory”, says Wood, so there is a strong incentive to develop better delivery mechanisms.
“In practice, resolving the chemistry and optimising the delivery has just taken longer to achieve [than for ASOs].”
Alnylam Pharmaceuticals has pioneered the siRNA approach and, in 2021, it submitted a clinical trial application in collaboration with Regeneron Pharma to the Medicines and Healthcare products Regulatory Agency for a phase I study of ALN-APP, an siRNA to treat early-onset Alzheimer’s disease by silencing the amyloid precursor protein (APP). The siRNA is designed to be delivered intrathecally in a single dose.
This will be Alnylam’s first siRNA programme using its new C16 conjugation technology to improve delivery to the brain. In addition to several modifications to the oligonucleotide backbone, the siRNA molecule is conjugated to a hexadecyl group. Exactly how this improves uptake in the brain is still being investigated.
“We hypothesise that it promotes moderate affinity interactions with the extracellular matrix and various cell membrane components, thus allowing for binding and uptake into various cell types within the brain as well as the lung and the eye,” explains Vasant Jadhav, head of the RNAi Platform at Alnylam Pharmaceuticals.
The company, based in Cambridge, Massachusetts, intends to use its promising siRNA approach to treat other neurodegenerative diseases.
There are a number of genetically defined neurodegenerative diseases where there is significant potential for CNS-targeted RNAi therapeutics to have an impact,
Vasant Jadhav, head of the RNAi Platform at Alnylam Pharmaceuticals
“We have development candidates that are undergoing preclinical evaluation for the potential treatment of Huntington’s disease and ALS in collaboration with Regeneron. We certainly feel that there are a number of genetically defined neurodegenerative diseases where there is significant potential for CNS-targeted RNAi therapeutics to have an impact,” says Jadhav.
Third-generation drugs
Now a third generation of gene-silencing drugs for delivery to the CNS are making their way to the clinic. A novel design comes from the research of Anastasia Khvorov at the University of Massachusetts and is now being developed through start-up company, Atalanta Therapeutics, also based in Massachusetts. They are developing a new chemically modified siRNA architecture called divalent siRNA (di-siRNA), which is made up of two parallel modified oligonucleotides linked together[12].
di-siRNA may be an improvement over both ASO and over ordinary siRNA, in terms of distribution, durability and tolerability
Alicia Secor, chief executive of Atalanta Therapeutics
So far, according to Alicia Secor, chief executive of Atalanta Therapeutics, their performance “suggests that di-siRNA may be an improvement over both ASO and over ordinary siRNA, in terms of distribution, durability and tolerability”.
Atalanta is collaborating with Biogen on an HTT-targeting treatment for Huntington’s disease, and with Genentech, based in San Francisco, California, to develop di-siRNA therapeutics for neurodegenerative diseases, including targets associated with Parkinson’s and Alzheimer’s, plus developing its own pipeline.
Another ‘third-generation’ approach comes from start-up Ceptur Therapeutics, based in New Jersey, which has attached U1 adapters to siRNA molecules. These short single strands of artificial RNA bind to the protein U1 snRNP — part of the machinery that modifies RNA molecules inside the nucleus after they are made. In this case, the adapters prevent the addition of the polyA tail stretch of the mRNA molecule, which is needed for translation — without it the nascent RNA is degraded. Ceptur recently completed a US$75m financing round to continue developing its therapeutic for Huntington’s disease.
Gene-silencing therapies based on siRNA are also now being delivered as gene therapies, using an adeno-associated viral vector to deliver the gene that makes the therapeutic RNA molecule.
Dutch biotech company, uniQure biopharma, is working on this approach to treat Huntington’s disease with its gene therapy candidate, AMT-130. The therapy delivers the gene directly to the caudate and putamen — two deep brain structures that are affected in people with Huntington’s disease, where information is processed. This will then continually produce the engineered RNA molecule, which will bind any mutated HTT protein and mark it for destruction. The procedure is done by catheter, guided by MRI. The first two patients in Europe have been dosed in uniQure’s phase I/II clinical trial, which is underway in Poland and is currently expanding to the UK[13]. “It’s a pretty invasive approach, high risk, [but] high reward,” says Wild, who is part of the University College London and Cardiff clinical trials team.
Future goals
Ultimately the ambition of many of those developing drugs for neurodegenerative conditions is to be able to edit the faulty genes causing the disease. Wood is confident that we will have the ability to do this in time. “I have absolutely no doubt this approach is going to work,” he says.
The current CRISPR technologies for gene editing are in their infancy and there are concerns about the accuracy of the editing process but, according to Watt, other base editing technologies that may be safer are coming down the pipeline towards readiness for clinical studies.
For now, there are still hurdles in the development of gene-silencing drugs, particularly for neurodegenerative conditions. One concern is improving the design of clinical trials and how patients are recruited, says Wood. “It’s really hard to take patients in whom the disease has progressed, in whom there’s an inflammatory environment in the brain, and expect a positive result.”
He suggests trials should concentrate on treating groups at an earlier stage who have a better chance of responding to drugs. Also important is better markers to measure disease progression and trial success.
What is it you actually measure in the patients that tells you that drug is working for something like Alzheimer’s?
Matthew Wood, professor of neuroscience in the Department of Paediatrics at the University of Oxford
“What is it you actually measure in the patients that tells you that drug is working for something like Alzheimer’s? That’s quite tough,” says Wood, whose team is trying to develop more sensitive cognitive and memory tests, so that it will be simpler to assess the next generation of gene-silencing drugs on their way.
Of course, it is important to remember that even if the gene-silencing drugs are able to reach targets deep in the brain, “if the target is wrong, we will still fail,’” says Watts.
While Huntington’s disease can be linked to a single gene, the situation is less simple for Alzheimer’s disease. A number of recent failures of drugs that target amyloid plaques, including crenezumab in June 2022, developed by Genentech and Swiss biotech company AC Immune, have led to questions about this approach. As delivery technology improves, gene-silencing drugs may ultimately help provide an answer.
“It becomes easier to cleanly address the questions of disease hypothesis, [and discover] what [we] can silence that will help a patient,” says Watts.
- 1Alzheimer’s Society’s view on demography. Alzheimer’s Society. 2020.https://www.alzheimers.org.uk/about-us/policy-and-influencing/what-we-think/demography (accessed 29 Jul 2022).
- 2Common forms of dementia. World Health Organization. 2021.https://www.who.int/news-room/fact-sheets/detail/dementia#:~:text=Common%20forms%20of%20dementia,60%2D70%25%20of%20cases. (accessed 29 Jul 2022).
- 3Parkinson’s diagnoses set to increase by a fifth by 2025. Parkinson’s UK. 2018.https://www.parkinsons.org.uk/news/parkinsons-diagnoses-set-increase-fifth-2025 (accessed 29 Jul 2022).
- 4Huntington’s In Mind, a campaign to highlight the mental health struggles associated with Huntington’s disease. Huntington’s Disease Association. 2022.https://www.hda.org.uk/news/huntington-s-in-mind-a-campaign-for-huntingtons-awareness-month (accessed 29 Jul 2022).
- 5Mummery CJ, Junge C, Kordasiewicz HB, et al. Results of the first‐in‐human, randomized, double‐blind, placebo‐controlled phase 1b study of lumbar intrathecal bolus administrations of antisense oligonucleotide (ISIS 814907; BIIB080) targeting tau mRNA in patients with mild Alzheimer’s disease. Alzheimer’s & Dementia. 2021;17. doi:10.1002/alz.051871
- 6Biogen. A Study to Assess the Efficacy, Safety, and Tolerability of BIIB080 in Participants With Mild Cognitive Impairment (MCI) Due to Alzheimer’s Disease (AD) or Mild Alzheimer’s Disease Dementia (CELIA). ClinicalTrials.gov. 2022.https://clinicaltrials.gov/ct2/show/NCT05399888 (accessed 29 Jul 2022).
- 7FDA Accepts Biogen’s New Drug Application and Grants Priority Review of Tofersen for a Rare, Genetic Form of ALS. BioSpace. 2022.https://www.biospace.com/article/releases/fda-accepts-biogen-s-new-drug-application-and-grants-priority-review-of-tofersen-for-a-rare-genetic-form-of-als/ (accessed 29 Jul 2022).
- 8Wave Life Sciences Provides Update on Phase 1b/2a PRECISION-HD Trials. Wave Life Sciences. 2021.https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-provides-update-phase-1b2a-precision-hd (accessed 29 Jul 2022).
- 9Ionis Pharmaceuticals. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ISIS 443139 in Participants With Early Manifest Huntington’s Disease. ClinicalTrials.gov. 2019.https://clinicaltrials.gov/ct2/show/NCT02519036 (accessed 29 Jul 2022).
- 10Roche provides update on tominersen programme in manifest Huntington’s disease. Roche. 2021.https://www.roche.com/media/releases/med-cor-2021-03-22b (accessed 29 Jul 2022).
- 11Mai-Lise N. Roche-Genentech HD Community Letter . Huntington’s Disease Society of America. 2022.https://hdsa.org/wp-content/uploads/2022/01/Roche-Genentech-HD-Global-Community-Letter-January-2022.pdf (accessed 29 Jul 2022).
- 12Alterman JF, Godinho BMDC, Hassler MR, et al. A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nat Biotechnol. 2019;37:884–94. doi:10.1038/s41587-019-0205-0
- 13UniQure Biopharma. Safety and Efficacy of AMT-130 in European Adults With Early Manifest Huntington Disease. ClinicalTrials.gov. 2022.https://clinicaltrials.gov/ct2/show/NCT05243017 (accessed 29 Jul 2022).