


This content was published in 2010. We do not recommend that you take any clinical decisions based on this information without first ensuring you have checked the latest guidance.
Summary
Epilepsy is not a specific disease but a symptom of a chronic, underlying neurological disorder. It is characterised by recurrent (two or more) seizures.
Diagnosis involves obtaining an accurate and detailed history from the patient (and where possible an eyewitness) of the events before, during and after a seizure took place. The intention is to identify the seizure type, any underlying causes or the presence of an epilepsy syndrome and any comorbidities.
Epilepsy (from the ancient Greek word epilepsia — meaning “to seize”) is the most common chronic neurological condition in the UK. An estimated 50 million individuals worldwide suffer from the condition and most of these people live in developing countries where diagnosis and treatment are under-resourced. Across age ranges, there is a bimodal distribution with the highest prevalence among the very young and the elderly.
The prevalence of epilepsy in England and Wales in 1998 was approximately 7.7 cases per 1,000 men and 7.6 cases per 1,000 women. The total annual cost of epilepsy to the governments in England and Wales (including direct and indirect costs) was estimated to be £2bn in 1992/93.1
The effect of epilepsy on quality of life varies; many people continue to function normally while others experience significant disability and social exclusion. The National Institute for Health and Clinical Excellence recommends that epileptic patients and their carers should be provided with information and education about all aspects of epilepsy, preferably with the aid of structured self-management plans.1
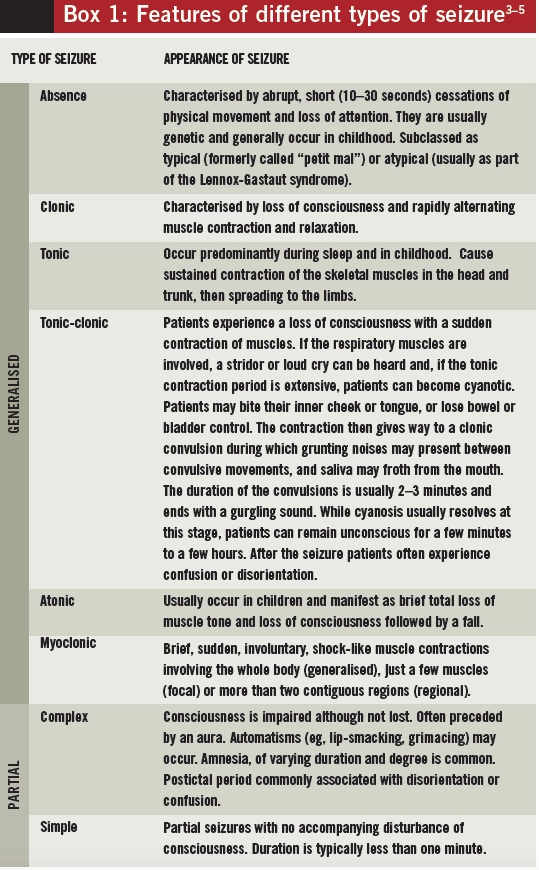
Seizure classification
A seizure is the manifestation of synchronous and abnormally excessive activity of neurons in the brain. The clinical presentation of a seizure might include changes in consciousness and behaviour as well as abnormal motor, sensory, autonomic or cognitive function.4 This presentation can vary depending on the part of the brain affected by the abnormal activity, the pattern of spread of neuronal discharge, the underlying cause and the age of the individual.
Occasionally, an aura is experienced before a seizure occurs. Fear, déjà vu, a rising feeling in the stomach or abnormal smells are among the sensations that can herald the onset of a seizure.
Partial (or focal) seizures are those for which the aberrant neuronal activation originates and remains localised in a specific area of the brain. They are further classified into:
- Complex — those that cause an impaired state or a loss of consciousness
- Simple — those that do not impair consciousness
The signs and symptoms of partial seizures depend on the cortical regions involved. Motor symptoms depend on the site of origin of the seizure. Most partial seizures involve one to two minutes of fitting (the ictus) but include longer postictal periods during which neurological dysfunction (eg, disorientation, memory problems) persists.
Generalised seizures are those where the aberrant neuronal activity is more widespread. They can be either primarily generalised (with onset occurring in both hemispheres of the brain simultaneously) or secondarily generalised (a partial seizure with a focal origin that spreads throughout the cortex). Consciousness is usually lost during generalised seizures and convulsions can, but do not always, occur.
A summary of the clinical features of different types of seizures is provided in Box 1. Seizures that do not fit into any category, often because an adequate description of events is not available, are known as unclassified seizures.
Provoked seizures
Seizures can be provoked or unprovoked. Those occurring as isolated events, where a systemic, toxic or metabolic brain injury is identifiable, are known as provoked seizures. They can occur as a result of various external factors, such as a head injury, stroke, uraemia or alcohol detoxification. By definition, provoked seizures do not recur if their cause is treated and does not recur. The likely causes of seizures vary according to a patient’s age when he or she experiences the first seizure. The common causes are summarised in Box 2.2

Prolonged and serial seizures
Convulsive seizures that last five minutes or longer are defined as “prolonged” whereas if three or more seizures occur within the space of an hour these are known as “serial seizures”. Each condition requires emergency treatment. Status epilepticus is a condition during which seizure activity persists for 30 minutes or longer, causing a wide spectrum of clinical symptoms.6 Generalised status epilepticus is a life-threatening medical emergency and rapid diagnosis and treatment are vital (see accompanying article, p89).
Epilepsy
Epilepsy is not a specific disease but a symptom of a chronic, underlying neurological disorder. It is characterised by recurrent (two or more) epileptic seizures not provoked by any immediately identifiable cause. The classification of epileptic syndromes and seizure types is somewhat discordant. However, an accurate diagnosis of epileptic syndrome is relevant to guide treatment choice, investigations, counselling and prognosis.
Epilepsy is not a unique single illness but often considered to be a group of syndromes (known as “the epilepsies”).1 Partial and generalised epilepsies can further be classified as:2,5
- Idiopathic — the origin is considered genetic
- Symptomatic — there is a known cause (eg, a tumour or lesion)
- Cryptogenic — signs, symptoms and diagnostic results indicate the presence of an as-yet unidentified underlying cause
The severity and prognosis of the condition vary according to the type of epilepsy.
Children and young people
Around 70% of people who develop epilepsy are diagnosed before the age of 20 years. In children, epilepsy is often identified as a syndrome even though it is not possible to establish which syndrome in up to 30% of cases. Serial seizures and status epilepticus (see below) are also more common in children.
Some common epileptic syndromes in children and young people include:4
- Benign childhood epilepsy with centrotemporal spikes — usually arises between seven and nine years of age but often remits before puberty
- Juvenile myoclonic epilepsy (JME) — onset usually occurs between 10 and 16 years of age and the condition frequently continues into adulthood. Approximately 50% of people with JME suffer from photosensitivity, where seizures are provoked by flashing lights (often from a television)
- Childhood absence epilepsy — presents as brief and frequent absence seizures (up to 100 per day) followed by abrupt loss of consciousness and may be associated with learning disabilities. Permanent remission often occurs at puberty but absences sometimes continue into adulthood and can progress to tonic-clonic or myoclonic seizures
- West syndrome — generalised epilepsy that usually starts in patients aged four to six months and is often associated with lifelong learning disabilities and neurophysiological abnormalities. The condition itself usually resolves by the time the patient reaches three years of age but up to half of affected infants go on to develop other epileptic syndromes, such as Lennox-Gastaut syndrome
- Lennox-Gastaut syndrome — causes frequent seizures of several seizure types. Most affected children develop severe learning disabilities that continue into adulthood
Elderly
Elderly people are more likely to develop seizures either provoked by acute illness or without an obvious precipitating cause. The annual incidence of epilepsy (recurrent unprovoked seizures) increases from 85.9 per 100,000 people aged 65–69 years to over 135 per 100,000 in those over 80 years of age.7
Elderly people with epilepsy have two to three times greater mortality than those without epilepsy.8 Most seizures are caused by focal area damage to the brain — the result of a stroke, a tumour, injury or a degenerative illness. Seizures that begin late in life are also a predictor of subsequent stroke.
Diagnosis
Epilepsy is difficult to diagnose and misdiagnosis is common. The UK Joint Epilepsy Council estimates that 23% of cases are misdiagnosed. For this reason, diagnosis should always be made by a specialist in epilepsy, defined by NICE as “a practitioner (or a paediatrician in the case of children) with training and experience in epilepsy”. Any person who experiences his or her first seizure should be seen by a specialist within two weeks to ensure an accurate diagnosis and initiation of therapy if appropriate.
Diagnosis involves obtaining an accurate and detailed history from the patient (and where possible from an eyewitness) of the events before, during and after a seizure took place. The intention is to identify the seizure type, any underlying causes or the presence of an epilepsy syndrome and any comorbidities. Syncope, drop-attacks, hypoglycaemia, some sleep disorders (eg, restless legs syndrome) and cardiac arrhythmias can all, initially, be mistaken for epilepsy.6
Investigations
Electroencephalography (EEG) can be helpful in determining diagnosis and classification of seizure type and syndrome. However, it should only be used to support a diagnosis in patients whose clinical history suggests that their seizures constitute epilepsy. EEG should not be used solely to make a diagnosis, nor should it be used to exclude a diagnosis of epilepsy.
Magnetic resonance imaging (MRI) is the preferred neuroimaging technique because it is more sensitive and does not expose patients to radiation. It is used to identify lesions (ie, tumours, vascular malformations) that could be causing the epilepsy. Computed tomography scanning can be used when an MRI is contraindicated or if an urgent assessment of seizures is required.
An electrocardiogram and appropriate baseline blood tests, such as electrolytes, glucose and calcium, should also be performed to identify possible causes and any comorbidities.6
Prognosis
Patients with epilepsy have a two to three times higher risk of mortality than the general public. Contributing factors include accidents caused by seizures, adverse events during epilepsy treatment, the underlying cause for symptomatic epilepsy (eg, a brain tumour), suicide or sudden unexpected death in epilepsy (SUDEP).
An increased risk of SUDEP has been linked to poor seizure control (often as a result of drug resistance) and the occurrence of nocturnal seizures. It is important that individuals with epilepsy and their carers are provided with information on SUDEP and how to minimise the risk of it occurring.
References
- National Institute for Health and Clinical Excellence. The epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care. October 2004. www.nice.org.uk/cg20 (accessed 22 February 2010).
- Elger CE, Schmidt D. Modern management of epilepsy: a practical approach. Epilepsy & Behavior 2008;12:501–39.
- National Institute for Health and Clinical Excellence. Newer drugs for epilepsy in adults. March 2004. www.nice.org.uk/ta76 (accessed 22 February 2010).
- National Institute for Health and Clinical Excellence. Newer drugs for epilepsy in children. April 2004. www.nice.org.uk/ta79 (accessed 22 February 2010).
- Stein MA, Kanner AM. Management of newly diagnosed epilepsy: a practical guide to monotherapy. Drugs 2009;69:199–222.
- Scottish Intercollegiate Guidelines Network. Diagnosis and management of epilepsies in children and young people. March 2005. www.sign.ac.uk/ guidelines/fulltext/81/index.html (accessed 22 February 2010).
- Wallace H, Shorvon S, Tallis R. Age-specific incidence and prevalence rates of treated epilepsy in an unselected population of 2,052,922 and age-specific fertility rates in women with epilepsy. Lancet 1998;352:1970–3.
- Lhatoo SD, Johnson AL, Goodridge DH, et al. Mortality in epilepsy in the first 11 to 14 years after diagnosis: multivariate analysis of a long-term, prospective, population-based cohort. Annals of Neurology 2001;49:336–44.
You might also be interested in…
Palliative care: end-of-life medicines management
