

Shutterstock.com
After reading this article, you should be able to:
- Understand the effects of critical illness on the pharmacokinetics and pharmacodynamics of commonly used drugs;
- Understand how to personalise drug therapy in critical illness according to the physiochemical properties of medicines, patient factors, disease factors and extracorporeal factors;
- Apply a conceptual framework for dosing strategies in critically ill patients.
Critical illness induces pathophysiological changes that cause significant alterations to drug pharmacokinetics and pharmacodynamics (PK-PD)[1]. PK refers to how the body handles drugs in terms of absorption, distribution, metabolism and elimination; and PD refers to the concentration effect of the drug at its site of action. Additionally, therapies designed to replace or support failing organs, including extracorporeal membrane oxygenation (ECMO) or renal replacement therapies (RRTs), impact drug PK-PD. The overall picture in critical illness is considerable PK-PD heterogeneity, which is highly dynamic with significant inter- and intra-individual variation.
Critical care pharmacists are members of the multidisciplinary team that care for patients in critical care areas[2]. Critical care pharmacists must be able to understand PK-PD properties of drugs and apply this knowledge to review the appropriateness of treatment plans. The Adult Critical Care services specification, the 2021 Adult Critical Care ‘Getting it right first time‘ document, and ‘Surviving sepsis’ guidelines are publications that highlight the vital role critical care pharmacists play in improving patient outcomes[3–5].
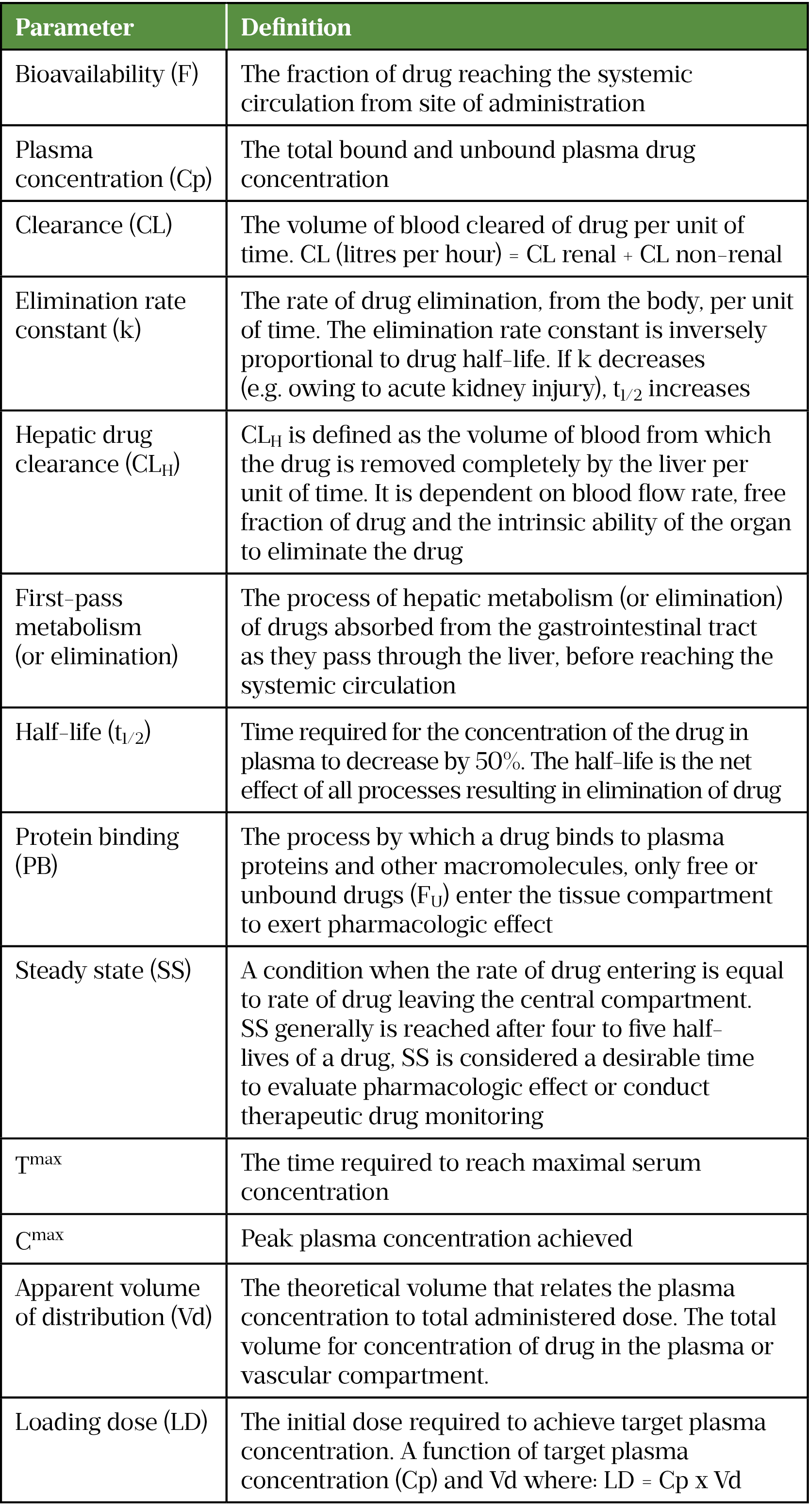
This learning article aims to outline PK-PD changes that occur in critical illness and how to optimise treatment while minimising harm. Common PK terms are described in Table 1[2].
Pharmacokinetics, pharmacodynamics and critical illness
Figure 1 describes the complex, dynamic interaction of critical illness-induced PK-PD changes that may increase or decrease plasma concentration[1]. Numerous drugs used to support patients during critical illness can be titrated to a PD target (e.g. sedative and analgesic agents titrated to sedation and pain scores, or cardiovascular medicines titrated to haemodynamic goals). However, for drugs that cannot be titrated to a PD goal, knowledge of PK properties, including volume of distribution (Vd), drug lipophilicity, elimination half-life and metabolism, should be applied to personalise pharmacotherapy.
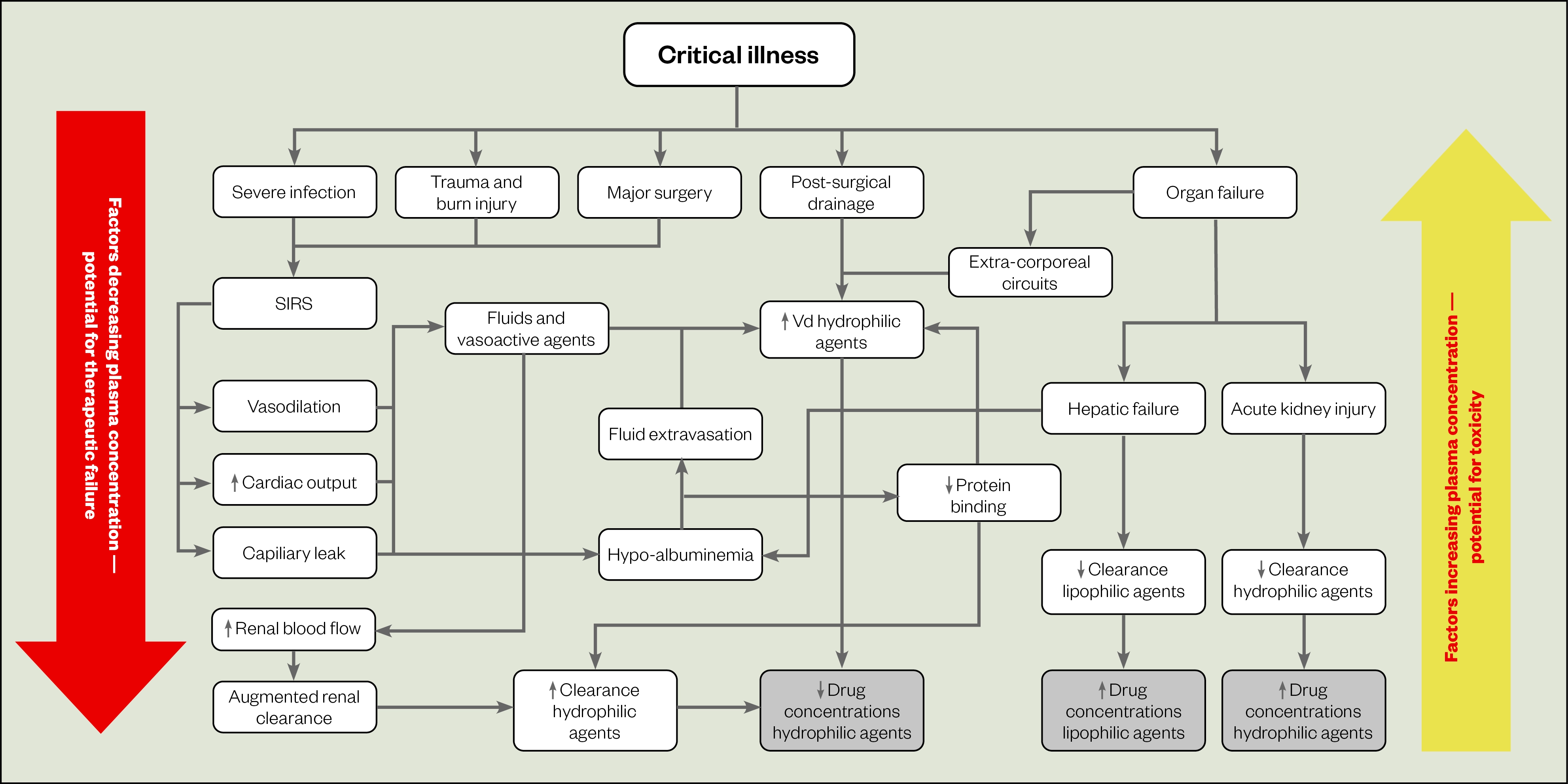
SIRS: Systemic inflammatory response syndrome
Source: Adapted from Blot et al.[1]
Absorption
Absorption of a drug occurs after enteral or oral administration, and is dependent on drug bioavailability (F). It is defined as the sum of fraction of drug absorbed (FA) minus the fraction eliminated, after first-pass elimination (FE)[6]. The fraction eliminated is the amount of drug removed by intestinal metabolism and efflux transporters (e.g. cytochromes P450, P-glycoprotein, multidrug resistance mutation 1), and hepatic first-pass elimination (or metabolism).
Intravenous (IV) administration is associated with a higher risk of error but is often preferred owing to unpredictability of oral/enteral absorption in critical illness. Breaks in feeding required for some enterally-administered medicines could result in non-attainment of nutritional targets and hypoglycaemia from unintended continuation of insulin therapy/antidiabetic medication. In addition, much of the literature for drug-feed interactions is not in a critical care context[7]. The risks and benefits of IV versus oral/enteral administration must be considered[2]. Table 2 describes pathological changes observed in critical illness that affect absorption and bioavailability[2,8].
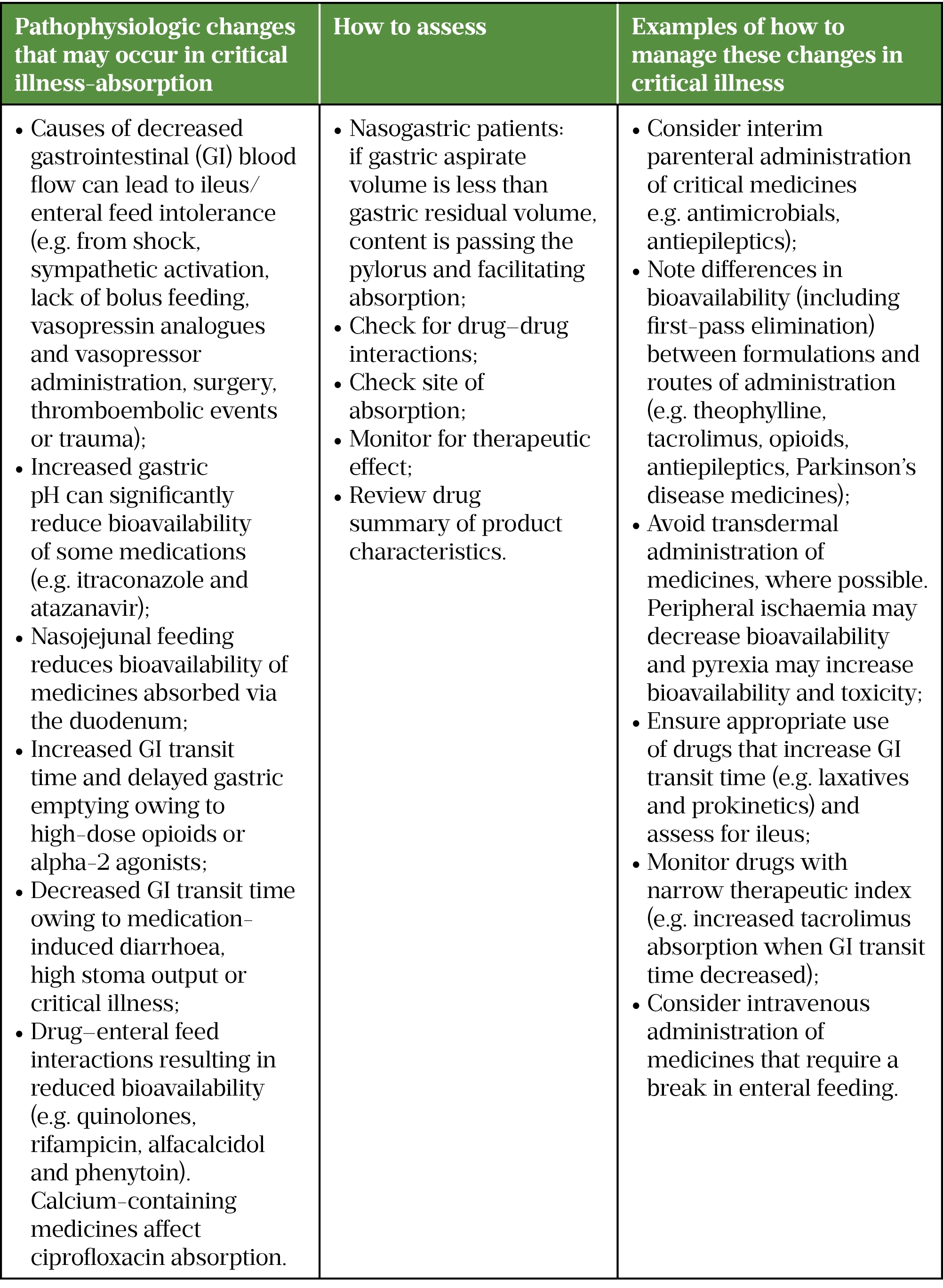
Sources: Critical Illness[9], Handbook of Drug Administration via Enteral Feeding Tubes, NEWT guidelines
Distribution
Drug distribution occurs immediately after absorption into the systemic circulation and is dependent on patient, disease, drug and treatment/extracorporeal-related factors. Several of these factors are affected by critical illness, including endothelial dysfunction contributing to increased Vd, altered protein binding, fluid resuscitation, organ dysfunction, fluctuating regional blood flow and cardiac output. Factors less affected by critical illness include patient factors (e.g. obesity, age) and drug factors (e.g. lipophilicity, partition coefficient, acid dissociation constant, degree of ionisation and site of elimination)[10].
Hydrophilic and lipophilic drugs
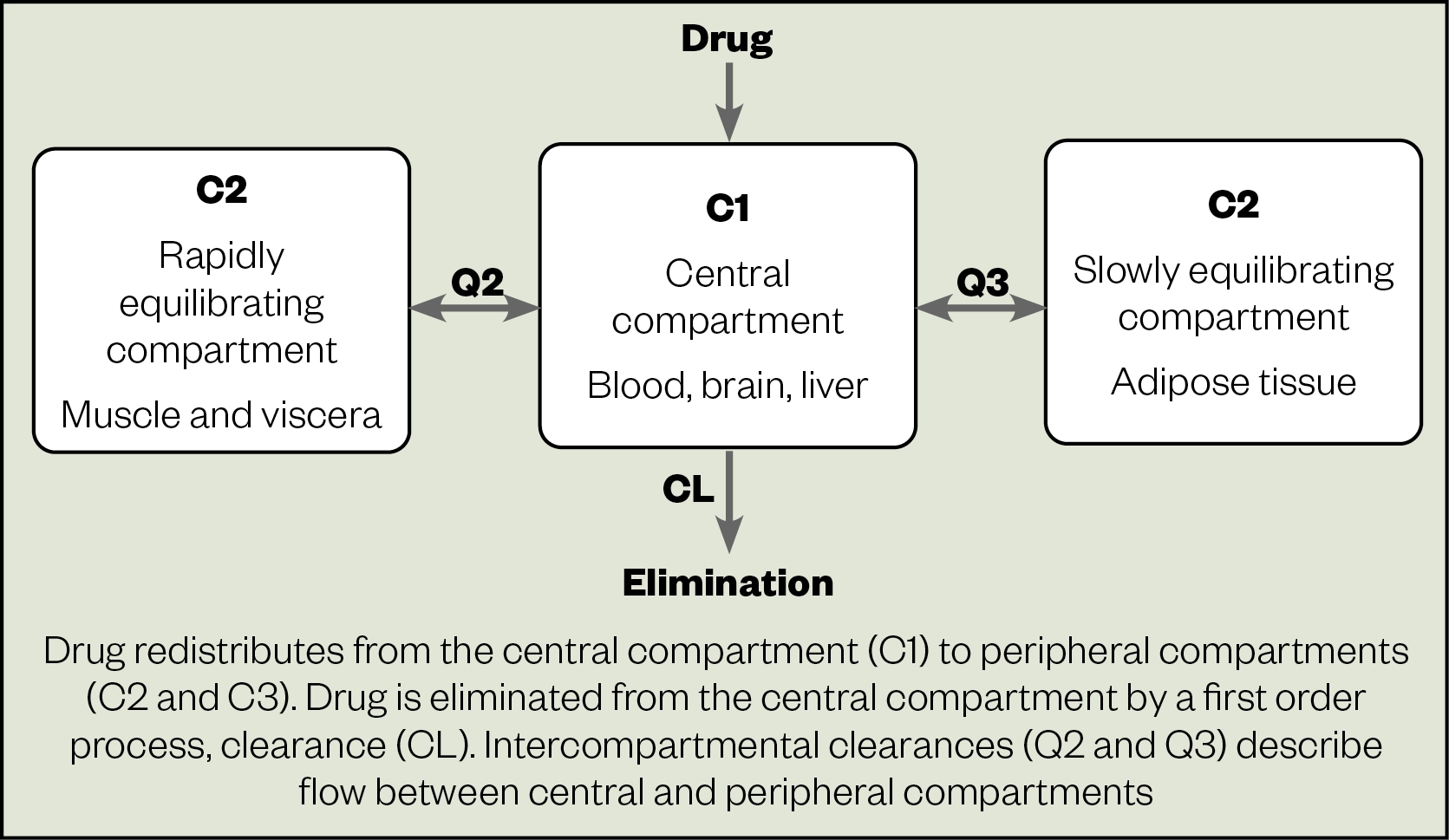
Distribution is influenced by a drug’s affinity for water (hydrophilic) or fat (lipophilic). Moderately lipophilic drugs, such as midazolam, distribute into a two-compartment model, the peripheral (tissue) compartment and the central (vascular) compartment. Highly lipophilic drugs (e.g. fentanyl, propofol and amiodarone) are described by a three-compartment model: the central/vascular compartment; fast-equilibrating well-perfused peripheral tissue compartment; and slower-equilibrating deep tissue/adipose with lower perfusion compartment (Figure 2). Hydrophilic drugs (e.g. beta-lactams, aminoglycosides and catecholamines) distribute in a single compartment (central/vascular), which may be affected by increases in Vd, as described below and in Table 3[10].
Increased Vd in critical illness may result in subtherapeutic drug plasma levels, with the potential for treatment failure and morbidity.
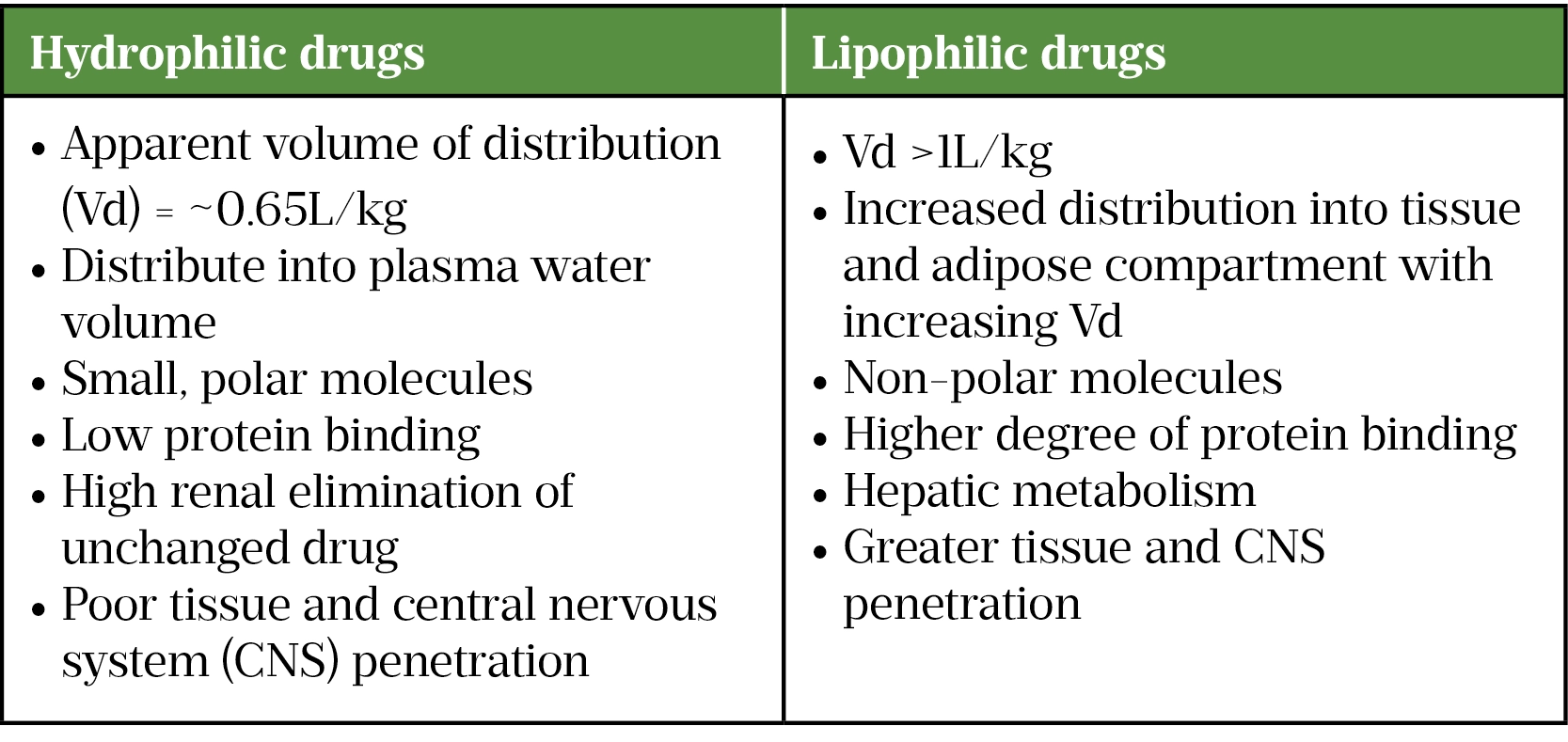
Hydrophilic drugs generally have poor tissue and central nervous system (CNS) penetration but this may increase when meninges are inflamed, or other transport mechanisms are present (e.g. active transport).
Sedatives and analgesics need to be lipophilic to penetrate the CNS and reach the site of action. Fentanyl and propofol have rapid tissue and CNS penetration, as well as rapid onset of action. At low dose/short infusions, plasma concentrations fall rapidly owing to rapid redistribution from tissue to plasma. With prolonged infusions, there is greater accumulation and concentration in peripheral (tissue and adipose) compartments, resulting in increased elimination half-life of lipophilic drugs. Drugs in the tissues/adipose can redistribute to the plasma compartment after cessation of infusion, leading to a prolonged, context-sensitive half-time[10].
The tissue/adipose compartments are relatively unchanged in critical illness, although reduced peripheral perfusion contributes to reduced distribution into these compartments during the early rescue phase of critical illness. Although some medicines are affected by this change, such as sedatives and opioids, they are titrated to pharmacodynamic goals. Conversely, the vascular compartment is significantly affected by critical illness-induced endothelial dysfunction. This results in capillary leak of intravascular fluid and albumin, contributing to tissue and organ oedema, impairing function. Additionally, vasodilation and fluid resuscitation increase Vd of hydrophilic drugs that primarily distribute into the vascular compartment. For example, the Vd of vancomycin is reported to double in critical illness[11].
![Table 4: Examples of changes in drug distribution in critical illness[9]](https://pharmaceutical-journal.com/wp-content/uploads/2022/02/Critical-illness-table-4.png)
Loading dose
Critically ill patients with septic shock or major burns can have increased Vd of the vascular compartment. In these cases, hydrophilic drugs will require a larger initial loading dose (LD), as described in Table 4, to achieve the same plasma concentration[11].
Protein binding
Hypoalbuminemia from sepsis or inflammation-induced endothelial dysfunction and fluid resuscitation can affect Vd for highly protein-bound drugs[12]. If the drug has a narrow therapeutic index, therapeutic drug monitoring (TDM) is advocated — for example, measuring free phenytoin levels or calculating the plasma level, corrected for hypoalbuminemia, to mitigate toxicity risk from increased unbound fraction (Fu) of drug. Conversely, α1-acid glycoprotein is an acute-phase plasma protein that is increased in critical illness and binds basic drugs (e.g. lidocaine); however, the consequence of this in critical illness is poorly understood[7].
CNS infection, neurological injury and hyperammonaemia can affect permeability of the blood-brain barrier, as well as drug efflux pump activity, potentially increasing toxicity for CNS-active drugs, such as opioids and benzodiazepines[13].
Obesity
Understanding the effects of obesity on drug dosing is imperative to mitigate risk of therapeutic failure or toxicity. Patients with a body mass index (BMI) greater than 30kg/m2 might require dose adjustment or mg/kg dosing (using adjusted or actual body weight) depending on the drug.
For hydrophilic agents, obesity has limited impact on Vd. Critical illness factors play a greater role in increasing Vd as described above. Small increases in Vd for hydrophilic drugs in obesity are owing to modest increases in blood volume and muscle mass. For lipophilic drugs, there will be increased Vd owing to distribution into adipose tissue. In practice, dosing of hydrophilic and lipophilic drugs based on a lean mass descriptor (e.g. adjusted body weight) is likely appropriate for obese patients[14].
Obesity patient factors and disease factors need consideration and TDM is encouraged[14]. In patients with low BMI, a mg/kg dosing regimen, using actual body weight, is recommended.
Extracorporeal therapies
Extracorporeal circuits (e.g. ECMO and CO2 removal) are used in specialist intensive care units (ICUs) to improve oxygenation, CO2 removal and/or cardiac function, when conventional ICU therapies have failed[15].
The additional crystalloid required for circuit priming increases hydrophilic drug Vd, potentially necessitating an increased LD[16]. Overall, ECMO has a smaller effect on hydrophilic drugs, and it is likely that pathophysiological factors have a greater impact on PK-PD than the ECMO circuit itself.
Both circuit tubing and membrane oxygenators have large surface areas that are reported to sequester lipophilic or highly protein-bound agents, including sedatives and opioids to varying degrees, increasing Vd and decreasing clearance[17]. These drugs are titrated to effect and should be tailored to patient and drug, although the impact of increased total drug exposure is not fully understood[18]. Continuous renal replacement therapy (CRRT) tubing can adsorb highly protein-bound drugs, including flucloxacillin, and particularly polyamide circuits[19,20]. TDM is advised where feasible.
Metabolism
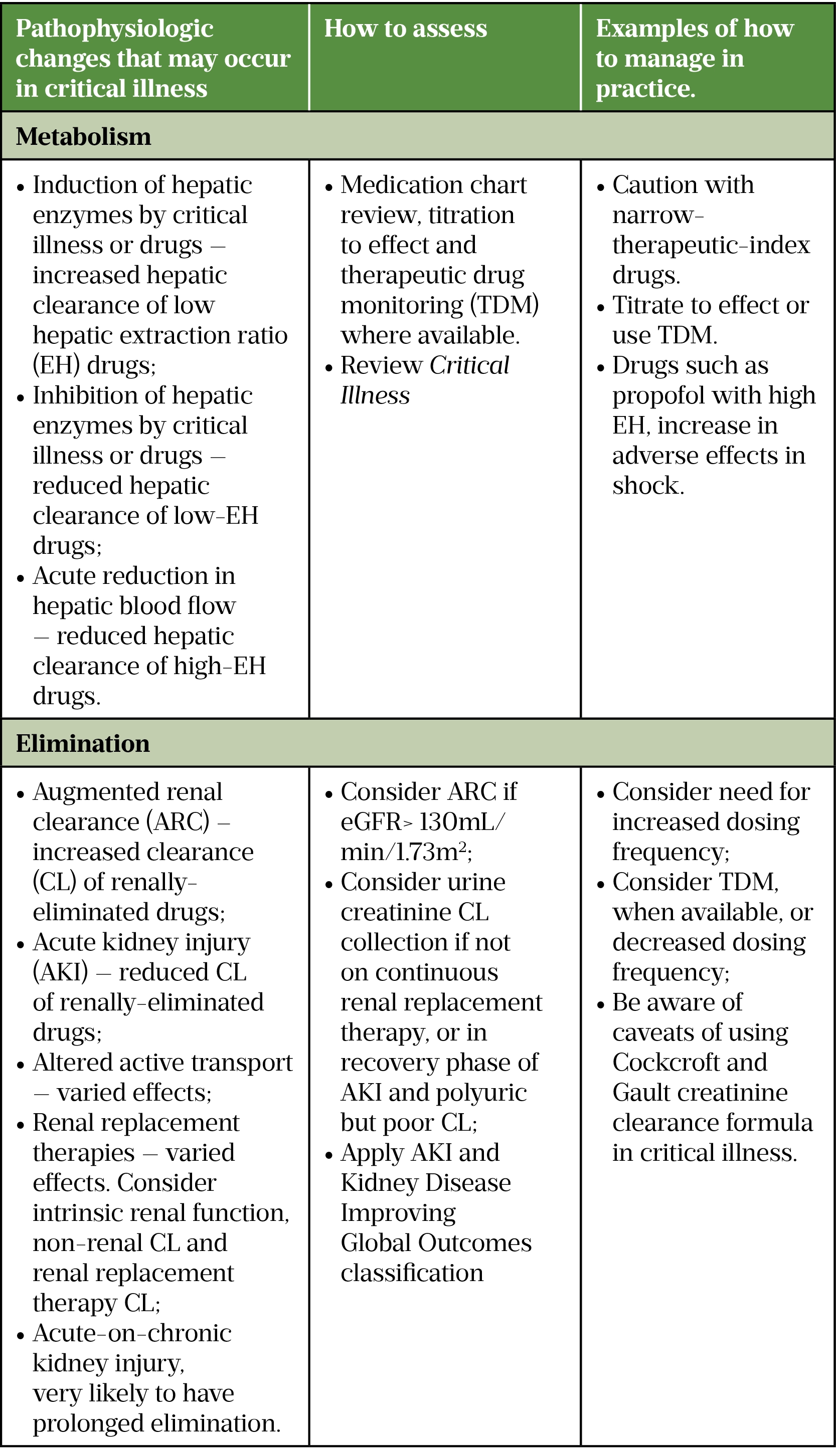
Renal and/or liver dysfunction is common in critical illness. These require daily pharmacist intervention to mitigate risks of accumulation and toxicity, or underdosing and treatment failure. For the majority of drugs, extrahepatic metabolism is non-significant (e.g. gut, lung, kidney, plasma esterases), with notable exceptions of remifentanil (plasma esterase) and atracurium/cisatracurium (Hofmann elimination)[10]. Clearance is defined as the volume of blood cleared per unit of time[2]. The determinants of drug clearance in most cases are hepatic metabolism and renal elimination.
Hepatic drug clearance (CLH), as defined in Table 1, is dependent on the fraction of Fu, the intrinsic enzymatic capacity of hepatocytes, known as hepatic extraction ratio, and hepatic blood flow (QH)[21].
The hepatic extraction ratio (EH) of drugs, as described in Table 5, is the fraction of drug cleared after a single pass through the liver. Drugs with high EH are rapidly cleared by the liver and dependent on hepatic blood flow. Therefore, positive pressure ventilation, vasopressors such as adrenaline (epinephrine) and critical illness-induced liver hypoperfusion may decrease liver blood flow and increase plasma levels of high EH drugs, with potential for toxicity (e.g. propofol and opioids). Increased propofol adverse effects, including decreased cardiac output and vasodilatation, have been reported with reduced QH, secondary to advanced heart failure or septic shock[22].
![Table 5: Hepatic extraction ratio of drugs used in critical care[10]](https://pharmaceutical-journal.com/wp-content/uploads/2022/02/Critical-illness-table-5.png)
Drugs with low EH are less dependent on liver blood flow and clearance is related to intrinsic metabolism by hepatocytes of unbound drug[10]. Highly protein-bound drugs (>90%) are less affected because of less fluctuation in plasma levels. Hepatic dysfunction induced by critical illness can include ischaemic hepatitis, indicated by transaminitis, which affects intrinsic metabolism, with the potential for accumulation and toxicity.
Intrinsic metabolism consists of phase 1 and phase 2 reactions. Phase 1 metabolism by cytochrome P450 includes hydrolysis, oxidation, dealkylation or reduction of molecules to produce a polar, hydrophilic metabolite. Phase 2 metabolism involves conjugation reactions to facilitate renal elimination of metabolites (e.g. morphine 6-glucluronide (M6G) and 1-hydroxymidazolam glucuronide (1OHMG)[23]. Therapeutic hypothermia and drug-drug interactions can affect enzymatic function with potential for reduced clearance (e.g. atypical antipsychotics used for delirium, proton pump inhibitors, macrolides, amiodarone, fluoroquinolones and azole antifungals) or increased clearance (e.g. rifampicin).
Excretion
Most drugs and metabolites are excreted or eliminated via the kidney. Acute kidney injury (AKI) is common in ICU, with an incidence of up to 50%[24]. The aetiology of AKI is multifactorial and beyond the scope of this article; however, renal blood flow is a major contributing factor.
Increased renal blood flow occurs in the early phase of sepsis owing to increased cardiac output. This can result in augmented renal clearance (ARC) as more drug is delivered to the kidney per unit of time[25]. ARC can necessitate higher or more frequent dosing of drugs that have high renal clearance (e.g. beta-lactams or aminoglycosides) to reduce risk of therapeutic failure.
More commonly, however, critically ill patients with AKI have reduced renal clearance. This may necessitate renal replacement therapy (RRT), typically delivered continuously in ICU. Renal replacement therapy clearance is dependent on prescribed and delivered dialysate and/or filtrate flow rates[26]. Only the unbound drug is removed by dialysis and filtration; this is often expressed as 1-PB,[27]. The greater the degree of protein binding, the lower the effect of RRT on clearance. The drug must be in the central compartment to be removed[26]. Dose adjustment in RRT is beyond the scope of this article.
The decision to reduce the drug dose/frequency required to support a patient in critical illness is equipoised between risks of accumulation and toxicity, and of therapeutic failure contributing to morbidity and mortality. For antimicrobials in sepsis and septic shock, the maximum tolerated dose, rather than minimum effective dose, should be considered as the consequences of underdosing may be significant. This is especially true for concentration-dependent antibacterial agents, including aminoglycosides[28].
The Renal Drug Database is an excellent source for patients with chronically impaired renal function, although some of the data is not validated in AKI and the PK-PD changes that occur in critical illness. The ‘Critical Illness‘ resource aims to account for these factors and advocates a daily, dynamic approach to drug dosing[9]. Additionally, the CRRT data in some PK-PD sources is prior to citrate anticoagulation: filter life was reduced and patients would experience time off RRT owing to circuit clotting. Table 6 describes factors to be considered when dosing critically ill patients with renal dysfunction.
Renal function
Assessment of renal function in critical illness is problematic for several reasons. Firstly, population-based equations used for glomerular filtration rate (GFR) such as the Chronic Kidney Disease Epidemiology Collaboration or the Cockcroft and Gault formula creatinine clearance are validated in stable chronic kidney disease, in which serum creatinine (SCr) excretion is at a steady state from muscle breakdown[29]. Dynamic changes in renal function, alongside unpredictable SCr excretion, render SCr an unreliable marker for assessing renal function in critical illness. Secondly, SCr is influenced by extremes of age, body weight, frailty, burns, or critical-illness-induced myopathy making SCr based equations unreliable[30].
Renal function assessment should be assessed on multiple parameters that give an overall indication of renal function, (e.g. urine output, SCr, pH, urea, base excess and potassium). A single marker is likely to be unreliable. The Kidney Disease Improving Global Outcomes classification for AKI can be used to give an indication of AKI severity (Table 6)[29].
![Table 6: Kidney Disease Improving Global Outcomes (KDIGO) classification for acute kidney injury (AKI)[29]](https://pharmaceutical-journal.com/wp-content/uploads/2022/02/Critical-illness-table-6.png)
A 4–8 hour urinary creatinine clearance measurement is a shorter version of the 24-hour collection and is a compromise between a timely result and adequate collection time. It has utility when SCr is unreliable (e.g. frail, low body weight-patients with low SCr, who require longer courses of renally-cleared drugs with risk of accumulation, such as IV aciclovir or IV colistin). TDM is advocated for these narrow therapeutic drugs.
Tubular secretion and reabsorption are additional considerations (e.g. fluconazole usually undergoes tubular reabsorption, therefore increased doses in patients receiving CRRT are required)[31].
Intra- and inter-individual variation
Common ICU syndromes, including sepsis and acute respiratory distress syndrome (ARDS), are heterogeneous syndromes rather than specific diseases. Significant inter-individual heterogeneity has been reported in sepsis and ARDS, with differing rates of organ dysfunction and inflammatory response[32,33]. This variation will likely result in heterogeneity in PK-PD, with the potential for differing responses to drug treatments. Stratification by common pathobiological mechanisms (endotypes), or clinical features (subphenotypes), could identify subgroups that may benefit from, or be harmed by, a treatment[34]. Steroid response in ARDS has been reported to vary depending on proposed hyper- and hypo-inflammatory phenotypes[35]. This is an emerging field in critical care and more data is needed if we are to achieve the goal of precision medicine where pharmacotherapy is tailored to person and pathology.
Additionally there is variation within a patient[1]. The host response changes over time with pro- and anti-inflammatory mechanisms dominating at different time points, resulting in varying effects on organ dysfunction and, in turn, drug PK-PD[36]. Additionally, organ dysfunction and recovery vary over time, often with second, or multiple, insults because of critical illness, further affecting the variation in PK-PD that occurs during a patient’s critical care stay.
Assessing drug therapy in critical illness
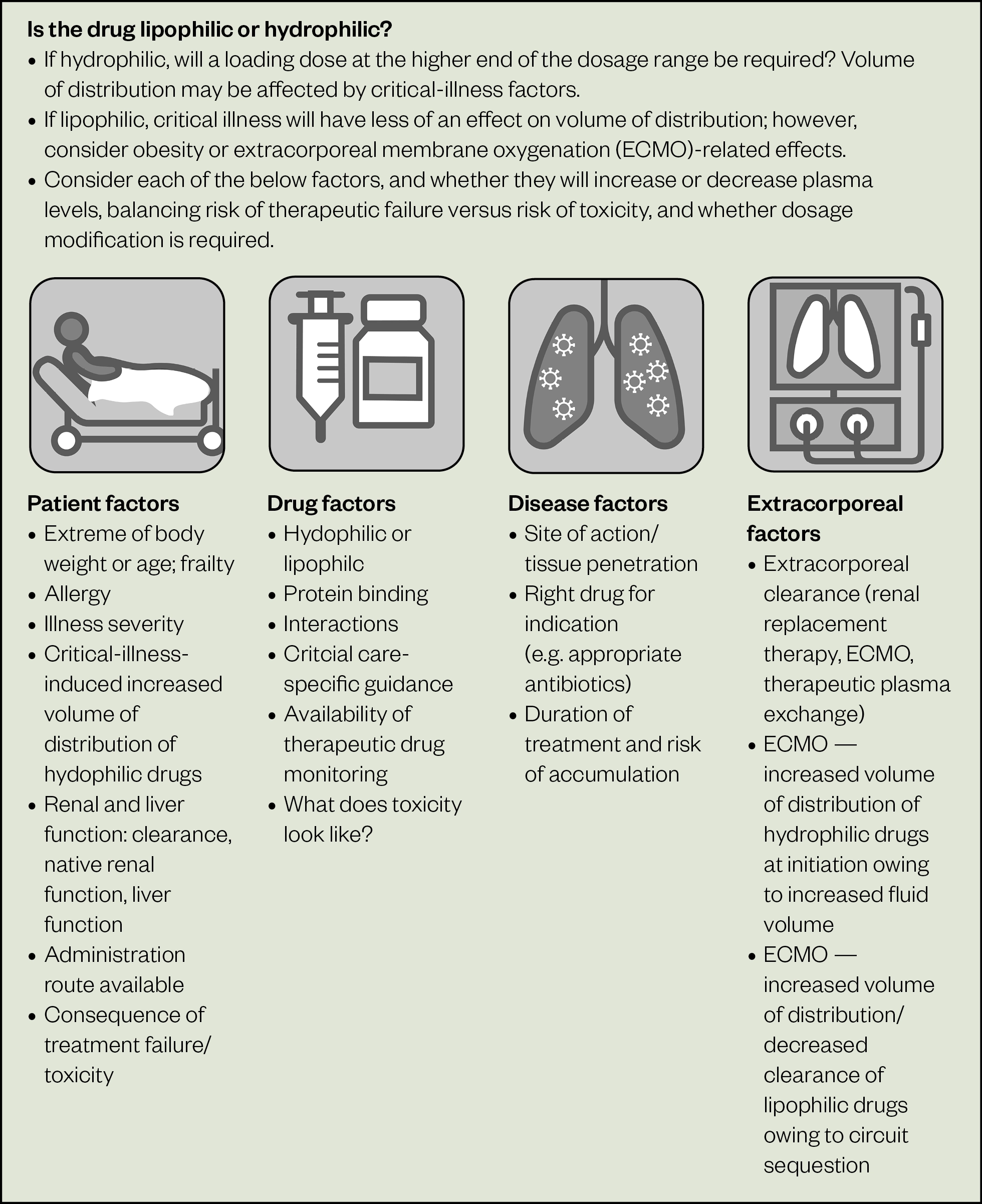
A conceptual framework can be used to assess the effects of critical illness on drug PK-PD and whether a change to pharmacotherapy is recommended, as summarised in Figure 3.
Case example
A young patient in septic shock, develops a significant hyperinflammatory response. In the initial phase of disease (hours), they may exhibit increased cardiac output to maintain blood pressure and organ perfusion, in response to sepsis-induced endothelial dysfunction and activation of the sympathetic nervous system, resulting in vasodilation. This inflammatory response damages endothelial glycocalyx, leading to increased vascular permeability, and results in decreased circulating volume and loss of intravascular fluid to the interstitial space[37].
Additionally, tissue and organ oedema contribute to organ dysfunction. To compensate for pathophysiological changes, the patient further increases cardiac output in response to vasodilation and endothelial dysfunction, to maintain organ perfusion and oxygen delivery. This increased cardiac output can lead to increased delivery of drugs to the clearance organs (mainly liver and kidney), with the sum effect of reduction in plasma concentration.
Over time, the haemodynamic compensation can become overwhelmed, and the patient develops distributive shock, which, if not treated quickly, can progress to multi-organ dysfunction and death. This organ dysfunction reduces clearance, which in turn increases plasma concentration. The challenge for the pharmacist is to balance the factors that affect plasma concentration and tailor pharmacotherapy for the patient.
Conclusion
Pathophysiological changes occur in critically ill patients that affect the PK-PD of drugs. These changes and their effect on management require daily review and reassessment by the pharmacist[38].
Understanding fundamental PK principles and the effects of critical illness will ensure safe and effective pharmacotherapy in the acutely unwell patient.
- 1Blot SI, Pea F, Lipman J. The effect of pathophysiology on pharmacokinetics in the critically ill patient — Concepts appraised by the example of antimicrobial agents. Advanced Drug Delivery Reviews. 2014;77:3–11. doi:10.1016/j.addr.2014.07.006
- 2Smith BS, Yogaratnam D, Levasseur-Franklin KE, et al. Introduction to Drug Pharmacokinetics in the Critically III Patient. Chest. 2012;141:1327–36. doi:10.1378/chest.11-1396
- 3Adult Critical Care services. NHS England. 2021.https://www.england.nhs.uk/publication/adult-critical-care-services/ (accessed Feb 2022).
- 4Evans L, Rhodes A, Alhazzani W, et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock 2021. Critical Care Medicine. 2021;49:e1063–143. doi:10.1097/ccm.0000000000005337
- 5Getting It Right First Time. FutureNHS. 2021.https://future.nhs.uk/system/login?nextURL=%2Fconnect%2Eti%2FGIRFTNational%2Fview%3FobjectId%3D112034405 (accessed Feb 2022).
- 6Abuhelwa AY, Williams DB, Upton RN, et al. Food, gastrointestinal pH, and models of oral drug absorption. European Journal of Pharmaceutics and Biopharmaceutics. 2017;112:234–48. doi:10.1016/j.ejpb.2016.11.034
- 7Roberts DJ, Hall RI. Drug absorption, distribution, metabolism and excretion considerations in critically ill adults. Expert Opinion on Drug Metabolism & Toxicology. 2013;9:1067–84. doi:10.1517/17425255.2013.799137
- 8Lemahieu W, Maes B, Verbeke K, et al. Cytochrome P450 3A4 and P-glycoprotein Activity and Assimilation of Tacrolimus in Transplant Patients with Persistent Diarrhea. Am J Transplant. 2005;5:1383–91. doi:10.1111/j.1600-6143.2005.00844.x
- 9Critical Illness. MedicinesComplete. 2022.https://about.medicinescomplete.com/publication/critical-illness/ (accessed Feb 2022).
- 10Charlton M, Thompson JP. Pharmacokinetics in sepsis. BJA Education. 2019;19:7–13. doi:10.1016/j.bjae.2018.09.006
- 11Vancomycin. MedicinesComplete. 2021.https://www.medicinescomplete.com/#/content/critical/106?hspl=vancomycin%23content%2Fcritical%2F106%23pharmacokinetics (accessed Feb 2022).
- 12Woodcock TE, Woodcock TM. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. British Journal of Anaesthesia. 2012;108:384–94. doi:10.1093/bja/aer515
- 13Zhang J, Zhang M, Sun B, et al. Hyperammonemia enhances the function and expression of P-glycoprotein and Mrp2 at the blood-brain barrier through NF-κB. J. Neurochem. 2014;131:791–802. doi:10.1111/jnc.12944
- 14Schetz M, De Jong A, Deane AM, et al. Obesity in the critically ill: a narrative review. Intensive Care Med. 2019;45:757–69. doi:10.1007/s00134-019-05594-1
- 15Allen S, Holena D, McCunn M, et al. A Review of the Fundamental Principles and Evidence Base in the Use of Extracorporeal Membrane Oxygenation (ECMO) in Critically Ill Adult Patients. J Intensive Care Med. 2011;26:13–26. doi:10.1177/0885066610384061
- 16Ha MA, Sieg AC. Evaluation of Altered Drug Pharmacokinetics in Critically Ill Adults Receiving Extracorporeal Membrane Oxygenation. Pharmacotherapy. 2017;37:221–35. doi:10.1002/phar.1882
- 17Cheng V, Abdul-Aziz M-H, Roberts JA, et al. Optimising drug dosing in patients receiving extracorporeal membrane oxygenation. J. Thorac. Dis. 2018;10:S629–41. doi:10.21037/jtd.2017.09.154
- 18Dzierba AL, Abrams D, Brodie D. Medicating patients during extracorporeal membrane oxygenation: the evidence is building. Crit Care. 2017;21. doi:10.1186/s13054-017-1644-y
- 19Meyer B, Ahmed el Gendy S, Delle Karth G, et al. How to Calculate Clearance of Highly Protein-Bound Drugs during Continuous Venovenous Hemofiltration Demonstrated with Flucloxacillin. Kidney Blood Press Res. 2003;26:135–40. doi:10.1159/000070997
- 20Li L, Li X, Xia Y, et al. Recommendation of Antimicrobial Dosing Optimization During Continuous Renal Replacement Therapy. Front. Pharmacol. 2020;11. doi:10.3389/fphar.2020.00786
- 21Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64:1147–61. doi:10.1007/s00228-008-0553-z
- 22HIRAOKA H. Changes in drug plasma concentrations of an extensively bound and highly extracted drug, propofol, in response to altered plasma binding. Clinical Pharmacology & Therapeutics. 2004;75:324–30. doi:10.1016/j.clpt.2003.12.004
- 23McKenzie CA, McKinnon W, Naughton DP, et al. Crit Care. 2005;9:R32. doi:10.1186/cc3010
- 24Case J, Khan S, Khalid R, et al. Epidemiology of Acute Kidney Injury in the Intensive Care Unit. Critical Care Research and Practice. 2013;2013:1–9. doi:10.1155/2013/479730
- 25Udy AA, Roberts JA, Shorr AF, et al. Augmented renal clearance in septic and traumatized patients with normal plasma creatinine concentrations: identifying at-risk patients. Critical Care. 2013;17:R35. doi:10.1186/cc12544
- 26Choi G, Gomersall CD, Tian Q, et al. Principles of antibacterial dosing in continuous renal replacement therapy. Critical Care Medicine. 2009;37:2268–82. doi:10.1097/ccm.0b013e3181aab3d0
- 27Pistolesi V, Morabito S, Di Mario F, et al. A Guide to Understanding Antimicrobial Drug Dosing in Critically Ill Patients on Renal Replacement Therapy. Antimicrob Agents Chemother. 2019;63. doi:10.1128/aac.00583-19
- 28Barclay ML, Begg EJ, Hickling KG. What is the Evidence for Once-Daily Aminoglycoside Therapy? Clinical Pharmacokinetics. 1994;27:32–48. doi:10.2165/00003088-199427010-00004
- 29Kellum JA, Lameire N. Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Critical Care. 2013;17:204. doi:10.1186/cc11454
- 30Hoste EAJ, Damen J, Vanholder RC, et al. Assessment of renal function in recently admitted critically ill patients with normal serum creatinine. Nephrology Dialysis Transplantation. 2005;20:747–53. doi:10.1093/ndt/gfh707
- 31Pittrow L, Penk A. Dosage adjustment of fluconazole during continuous renal replacement therapy (CAVH, CVVH, CAVHD, CVVHD). Mycoses. 1999;42:17–9. doi:10.1046/j.1439-0507.1999.00269.x
- 32Heijnen NFL, Hagens LA, Smit MR, et al. Biological Subphenotypes of Acute Respiratory Distress Syndrome Show Prognostic Enrichment in Mechanically Ventilated Patients without Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med. 2021;203:1503–11. doi:10.1164/rccm.202006-2522oc
- 33Seymour CW, Kennedy JN, Wang S, et al. Derivation, Validation, and Potential Treatment Implications of Novel Clinical Phenotypes for Sepsis. JAMA. 2019;321:2003. doi:10.1001/jama.2019.5791
- 34van der Poll T, Shankar-Hari M, Wiersinga WJ. The immunology of sepsis. Immunity. 2021;54:2450–64. doi:10.1016/j.immuni.2021.10.012
- 35Chen H, Xie J, Su N, et al. Corticosteroid Therapy Is Associated With Improved Outcome in Critically Ill Patients With COVID-19 With Hyperinflammatory Phenotype. Chest. 2021;159:1793–802. doi:10.1016/j.chest.2020.11.050
- 36Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. 2017;14:121–37. doi:10.1038/nrneph.2017.165
- 37Chelazzi C, Villa G, Mancinelli P, et al. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care. 2015;19. doi:10.1186/s13054-015-0741-z
- 38Barrett NA, Jones A, Whiteley C, et al. Management of long-term hypothyroidism: a potential marker of quality of medicines reconciliation in the intensive care unit†. International Journal of Pharmacy Practice. 2012;20:303–6. doi:10.1111/j.2042-7174.2012.00205.x
1 comment
You must be logged in to post a comment.



Excellent article