CNRI / Science Photo Library
In this article you will learn:
- How sarcoidosis can present
- The prognosis of patients with sarcoidosis
- Treatment options that may be considered in managing sarcoidosis
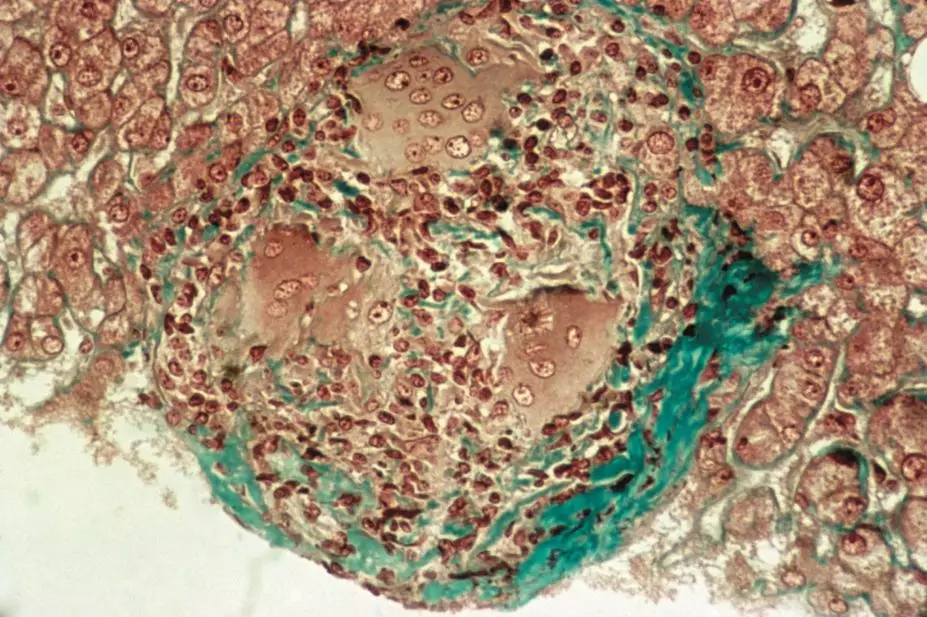
Sarcoidosis is a non-malignant immunological disorder characterised by non-caseating (non-necrotising) granulomas. These most commonly affect the lungs, eyes, lymph nodes and skin, although any tissue can be affected.
There is considerable variation in the incidence and prevalence of sarcoidosis around the world. This is probably explained by differences in environmental exposures and genetic background. For example, there is a higher incidence in some geographic areas, such as Scandinavia, and in some ethnic groups, such as African Americans.
In the UK, the incidence of sarcoidosis is around three to five cases per 100,000 per year[1]
. These cases can present at any age, with a peak age of onset between 25 and 40 years[2]
. Sarcoidosis is around 20% more common in women than men[2]
.
The organs affected in sarcoidosis and patients’ symptoms are variable. In some cases the condition may resolve completely without the need for intervention, while other patients may require prolonged treatment with immunosuppressants. The condition may be resistant to treatment, and long-term therapy may be required to reduce damage to the organs involved.
Pathophysiology
Granulomas are organised collections of activated macrophages, which form in response to certain antigens. It is likely that the development of sarcoidosis is dependent on the interplay and specific arrangement of the type of antigen, human leukocyte antigen (HLA) class II molecules, and T cell receptors.
Granulomas in sarcoidosis are similar to those seen in tuberculosis, and contain macrophages, monocytes and active T-lymphocytes. However, unlike tuberculosis, these are non-caseating and sarcoidosis does not appear to be the result of an active infection.
The antigens that can trigger the immune response in sarcoidosis are unknown. The reason why certain individuals develop sarcoidosis in response to them is probably explained by variations in the immune response in susceptible individuals. Given the lungs, eyes, skin and lymphatic system are commonly affected, the antigens causing sarcoidosis are thought to be environmental and sarcoidosis is not thought to be an autoimmune condition. Risk factors include variations in the HLA-DRB1 gene, which affects antigen presentation to T-lymphocytes, which appear to be a major factor in the development of sarcoidosis. Different HLA alleles have been shown to be associated with different disease phenotypes; for example, HLA-DRB1*03 and DQB1*0201 are associated with acute onset and resolving disease, whereas HLA-DRB1*15 and DQB1*0601 are associated with chronic disease[3]
.
Clinical features
Given the range of organs that can be involved, sarcoidosis may present in many ways and there is a potential for confusion with other conditions.
The lungs are involved in 80–90% of cases, and asymptomatic sarcoidosis is sometimes identified following a chest X-ray. Typical presenting symptoms include a dry cough, shortness of breath or chest pain. These non-specific respiratory symptoms can lead to a delay in diagnosis or a misdiagnosis of asthma or chronic obstructive pulmonary disease (COPD).
The development and worsening of interstitial fibrosis leading to respiratory failure is an uncommon but significant complication in pulmonary sarcoidosis and is a considerable cause of morbidity and premature mortality. Pulmonary fibrosis is also a significant risk factor for the development of aspergilloma and pulmonary hypertension[4]
.
Pulmonary hypertension occurs in 1–6% of cases of pulmonary sarcoidosis and can occur in the absence of significant pulmonary fibrosis. The main risk factors are the presence of pulmonary fibrosis and possibly male sex[5]
. Potential signs of pulmonary hypertension include breathlessness (out of proportion with the extent of pulmonary disease), palpitations, or feeling faint on exertion or leaning forward.
Enlarged lymph nodes, typically in the anterior triangle of the neck, occur in around one third of patients. Enlarged nodes caused by sarcoidosis will be painless and mobile, and can be misdiagnosed as possible lymphoma.
The eyes are affected in 10–80% of patients with sarcoidosis at some point during the course of the condition[6]
. The most common presenting symptom is uveitis, although any part of the eye or orbit may be affected. Uveitis is potentially sight-threatening and given that the majority of cases present insidiously without acute symptoms, periodic ophthalmological examination is recommended for patients with sarcoidosis. There are no guidelines or consensus recommendations, although most specialists recommend this occurs at least annually. Other symptoms associated with acute manifestations include pain and photophobia.
Skin lesions are common in people with sarcoidosis, and a range of non-specific lesions can occur[7]
. Two identifiable lesions associated with sarcoidosis are erythema nodosum and lupus pernio.
Erythema nodosum is an inflammatory skin disorder that commonly occurs in acute sarcoidosis, although it can occur in other conditions, such as streptococcal infections, inflammatory bowel disease or pancreatic cancer. It presents as raised, red, tender bumps or nodules on the anterior of the legs, most commonly the shins, and typically with symmetrical distribution. Erythema nodosum may be associated with systematic symptoms such as fever, malaise and arthralgia in nearby joints.
Erythema nodosum lesions are associated with inflammation and neutrophilic infiltration of subcutaneous fatty tissue, and the lesions are typically self-limiting. They progress from bright red to a brownish-yellow colour, and both old and new lesions may co-exist and look similar to bruising or cellulitis. Löfgren’s syndrome, an acute, benign and self-limiting form of sarcoidosis, is characterised by erythema nodosum and bilateral hilar lymphadenopathy (enlargement of lymph nodes in the pulmonary hila).
Lupus pernio is a rare manifestation of sarcoidosis that causes disfiguring skin lesions. It is more common in Afro-Caribbean women and presents as reddish-purple, nodular skin lesions, typically on the nose, cheeks, earlobes, lips and forehead. Lupus pernio is commonly associated with pulmonary fibrosis, chronic uveitis and sarcoidosis of the upper respiratory tract. Patients who develop lupus pernio often have chronic disease that does not respond to treatment.
Hypercalcuria occurs in around 30% of patients with sarcoidosis, and hypercalcaemia occurs in around 10% of patients[8]
. This is because granulomas can activate ergocalcitrol (vitamin D2) to calcitriol (vitamin D3) by producing 1-α hydroxylase. This can lead to an abnormal vitamin D2: vitamin D3 ratio. One study found that while 80% of sarcoidosis patients had low vitamin D2 levels, only 1% have low vitamin D3 levels and 10% have elevated vitamin D3 levels[9]
. As the vitamin D2 level can be disproportionately low in patients with sarcoidosis, the measurement of this alone (the standard laboratory assessment) may be misleading. The optimal blood concentration of vitamin D in a patient with sarcoidosis is unclear.
Both calcium and vitamin D supplements can cause acute renal failure in patients with sarcoidosis, and use should be considered on a case by case basis.
Sarcoidosis is an independent risk factor for fractures[10]
. This risk is increased in patients taking long-term oral corticosteroids. Patients with sarcoidosis are at increased fracture risk at a vitamin D2 level both below 10ng/ml and above 20ng/ml[11]
.
Neurosarcoidosis can affect almost any component of the central nervous system, although it most commonly affects the cranial nerves (particularly the facial and optic nerves). Symptoms of neurosarcoidosis include ataxia, cognitive dysfunction, headache, seizures and muscle weakness.
Rare manifestations of neurosarcoidosis include Heerfordt’s syndrome, which presents as facial palsy accompanied by fever, swelling of the parotic glands and uveitis; and granulomas in the pituitary gland, which can result in pituitary insufficiency and diabetes insipidus[12]
.
Haematological changes, such as decreased haemoglobin levels and platelet counts[13]
, can be caused by enlargement of the spleen. This occurs in 5–10% of patients with sarcoidosis, but is usually minimal and asymptomatic. The most common haematological abnormality caused by sarcoidosis is lymphopaenia, which is due to the relocation of circulating lymphocytes to the site of active disease.
The liver is affected in around 60–80% of cases, although clinical signs are less common and liver dysfunction (usually cholestasis) occurs in around 20% of patients with granulomas in the liver[8]
.
The heart can be affected at any time during the course of sarcoidosis. Although only a small number of patients are affected, cardiac complications can lead to sudden cardiac death and all patients diagnosed with sarcoidosis should be screened periodically using an electrocardiogram (ECG). There are no guidelines on how commonly this should occur, although most clinicians will screen patients at least once a year if there are cardiac signs or symptoms[9]
. Cardiac involvement is more common in patients of Japanese ancestry and in this group, heart failure and cardiac conduction abnormalities are the main cause of mortality.
Depression and neuropsychiatric illness often accompany sarcoidosis and concomitant depression has been reported to occur in up to two thirds of patients with sarcoidosis[5]
.
Nonspecific symptoms, such as fever, fatigue, malaise and weight loss, are common complications in patients with sarcoidosis. Significant fatigue affects up to 70% of patients and can be disabling, although other possible causes (e.g. diabetes) should be ruled out[2]
.
Diagnosis
Some presentations of sarcoidosis, Löfgren’s syndrome for example, are highly suggestive of sarcoidosis. For other presentations, the diagnosis can be difficult because of the wide range of presentations and overlap with several other diseases, necessitating confirmation of the diagnosis by a specialist service following extensive investigations. In complex cases, referral to a multidisciplinary specialist team in a tertiary centre may be required.
Sarcoid granulomas can be confirmed with histological samples obtained via biopsy, and support a diagnosis of sarcoidosis once other possible causes have been excluded.
Around 60% of patients with sarcoidosis have elevated serum angiotensin converting enzyme (sACE) levels, which can further support the diagnosis[8]
. The level of sACE does not appear to relate to the severity of disease, and has no role in the monitoring of patients with confirmed diagnosis. Patients should stop taking any ACE inhibitors before sACE levels are tested.
Bronchoalveolar lavage with collection of cells from the lungs can help differentiate sarcoidosis from other forms of interstitial lung disease. In patients with sarcoidosis, there is commonly an increased number of lymphocytes (>10–15% of cells), with a CD4+/CD8+ ratio >3.5[14]
.
Sarcoidosis often affects multiple organs, and patients with sarcoidosis confirmed in one organ should receive further tests as appropriate, including a chest X-ray, skin assessment, ophthalmic assessment, an ECG, neurological assessment, liver function test and evaluation of serum and urinary calcium levels.
Prognosis
The prognosis of sarcoidosis is variable. In some cases no treatment is required and spontaneous resolution occurs within one to three years. The risk of recurrence following spontaneous remission within two years is around 10%[15]
. Sarcoidosis will last for longer than three years in around 30% of patients[2]
. Sarcoidosis is fatal in less than 5% of patients and generally results from respiratory failure due to pulmonary fibrosis.
Patients with sarcoidosis that affects the respiratory system can be categorised following a chest X-ray using the Scadding system[16]
(see ‘Scadding system for assessing sarcoidosis’). Although this is a valid prognostic marker, it needs to be considered in the context of other organs involved, functional impairment and the patient’s symptoms[3]
.
| Scadding system for assessing sarcoidosis | ||
|---|---|---|
Stage | Radiological findings | Probability of resolution within two years |
I | Bilateral hilar lymphadenopathy | 50–90% |
II | Bilateral hilar lymphadenopathy with pulmonary infiltrations | 40–70% |
III | Pulmonary infiltrations without bilateral hilar adenopathy | 10–20% |
IV | Pulmonary fibrosis | 0% |
Other risk factors that reduce the likelihood of resolution include: Afro-Caribbean ethnicity; onset after 40 years of age; or the presence of lupus pernio, chronic uveitis, chronic hypercalcaemia, or disease involving the central nervous system or heart.
Management
In patients with a high chance of spontaneous resolution (i.e. Löfgren’s syndrome, or Scadding stage I disease limited to the lungs), no treatment may be required and the patient will instead be monitored regularly.
In patients with a low likelihood of spontaneous resolution (i.e. Scadding stage III or multiple organ involvement), or if there are significant symptoms or a high risk of irreversible organ damage (particularly to the central nervous system or heart), early treatment with systemic therapies is normally considered.
The World Association for Sarcoidosis and Other Granulomatous Disorders (WASOG) have produced guidelines to assist in treatment decisions[17]
. However, as the condition is variable, early involvement from a specialist with experience in the management of sarcoidosis of the affected organs is usually required.
In some situations, such as disease limited to the skin or anterior uveitis, topical therapy may be sufficient.
Systemic corticosteroids are the first-line treatment for the majority of patients with sarcoidosis. There are no randomised controlled trials evaluating the optimal dose or choice of corticosteroid, but in practice, treatment is generally started with prednisolone at a dose of 20–40mg daily. In patients with severe sarcoidosis symptoms (typically patients with cardiac, ophthalmological or central nervous system symptoms), up to 1mg/kg/day of prednisolone, or intravenous methylprednisolone 500mg–1g daily for three to five days, before switching to oral prednisolone, have been used.
Doses are normally maintained for four to twelve weeks and then assessed. If the patient has shown limited improvement, a corticosteroid-sparing agent (e.g. hydroxychloroquine, methotrexate, azathioprine) may be added, with the patient’s response reassessed after at least six to eight weeks of combined treatment.
If the patient shows improvement with prednisolone alone, the dose can be tapered by 5mg per day every two to four weeks, until a maintenance dose of 5–10mg prednisolone per day is achieved. If a maintenance dose of 10mg or less cannot be achieved without a recurrence or worsening of symptoms, a steroid-sparing agent should be considered as adjunct therapy.
For patients in whom the condition improves, the consensus is that a minimum of 12 months treatment is required. In some acute cases shorter durations of three to six months may be sufficient but further evidence is needed to identify patients for whom this approach may be appropriate.
Treatment with corticosteroids can be tapered and stopped if disease activity resolves. However, previous pharmacological treatment is one of the most significant predictors of disease recurrence, and up to 80% of patients treated with corticosteroids experience relapse in the first two years following tapering and discontinuation of treatment (compared to 10% in patients who achieve spontaneous remission)[15]
. Relapse is most common within the first six months of discontinuing therapy, and is rare after three years of remission[2]
. Patients are therefore typically followed up closely during the initial period after discontinuation, with prompt retreatment if there is recurrence. There is no fixed schedule for follow-up, although usually this will be every few months for the first year, with decreasing frequency until they are discharged from the service, usually after two years.
Side effects of treatment with oral corticosteroids include psychiatric disturbances, dyspepsia , increased risk of gastrointestinal bleeding, diabetes mellitus, adrenal suppression, increased susceptibility to infections, thinning of the skin, increased fracture risk, fluid retention and increased blood pressure.
Hydroxychloroquine has been used as an adjuvant to corticosteroids and alone in patients with mild sarcoidosis, typically at a dose of 200mg twice daily[17]
.
Ocular toxicity is associated with hydroxychloroquine, and patients should receive an eye examination every six to twelve months and will be advised to tell their GP about any changes in vision promptly, especially scotoma (a blind spot) while reading, diminished colour and night vision, or halos around lights.
Hydroxychloroquine may lower blood glucose levels and gastrointestinal intolerance is a relatively common side effect. Hydroxychloroquine may also precipitate haemolytic anaemia in patients with a deficiency in the enzyme Glucose-6-Phosphate Dehydrogenase (G6PD). This deficiency is more common in patients originating from the tropics and subtropics, and may be as high as 20% in some populations. G6PD activity can be assessed by a blood test and should be considered before starting treatment, particularly if the patient is of Mediterranean or African ethnicity.
Methotrexate is one of the most commonly used corticosteroid-sparing agents in sarcoidosis, typically at a dose of 5–15mg once weekly, although higher doses have been used. Methotrexate may be used as monotherapy in patients unable to take or tolerate corticosteroids, although it can take several months for a response. There are no clear data on efficacy, and treatment is typically assessed two to four months after initiation[18]
.
Folic acid is often given concomitantly with methotrexate to reduce side effects such as mucositis and gastrointestinal upset, typically at a dose of 5mg the day after methotrexate is taken.
Methotrexate is cleared renally and associated with haematological and hepatic toxicity. During treatment, patients will require blood tests, typically fortnightly full blood counts and liver function tests until six weeks after stabilisation of treatment, then monthly, along with an assessment of renal function every three months. Methotrexate is associated with a number of clinically significant interactions, and because of the risk of liver injury, patients must ensure they limit alcohol intake to well within recommended limits.
Azathioprine has been reported to be as effective as methotrexate in the treatment of sarcoidosis[17]
. However, its evidence base is more limited and there appears to be a higher incidence of opportunistic infections and a possible increased risk of malignancy compared with methotrexate. Azathioprine therefore tends to be reserved for patients in whom methotrexate is contraindicated.
Side effects of azathioprine include dyspepsia, oral ulcers, muscle pain, hepatic dysfunction, blurred vision and pancreatitis.
Azathioprine is metabolised by the enzyme Thiopurine S-MethylTransferase (TPMT), and TPMT activity should be assessed before starting treatment to identify patients at increased risk of toxicity or treatment failure.
Cytokine modulators are known to precipitate sarcoidosis in some patients. However, there is increasing evidence that infliximab and adalimumab may have a role in patients with chronic treatment-resistant sarcoidosis[19]
.
Other corticosteroid-sparing agents, including ciclosporin, cyclophosphamide, leflunomide, mycophenolate mofetil and pentoxifylline, have been used in difficult cases based on limited evidence. There is contradictory evidence, both for and against, the use of thalidomide in the management of lupus pernio.
Lloyd Mayers is a respiratory pharmacist and Sarah Mulholland MRPharmS is a rotational specialist clinical pharmacist, both at North Bristol NHS Trust.
References
[1] Gribbin J, Hubbard RB, Le Jeune I et al. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006;61:980–985.
[2] Valeyre D, Prasse A, Nunes H et al. Sarcoidosis. Lancet 2013;6736:1–13.
[3] Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011;183:573–581.
[4] Baughman RP, Lower EE, Gibson K. Pulmonary manifestations of sarcoidosis. Presse Med 2012;41:e289–302.
[5] Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA 2011;305:391–399.
[6] Bodaghi B, Touitou V, Fardeau C et al. Ocular sarcoidosis. Presse Med 2012;41:e349–354.
[7] Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med 2012; 41: e355–374.
[8] Nunes H, Bouvry D, Soler P et al. Sarcoidosis. Orphanet J Rare Dis 2007;2:46.
[9] Conron M, Young C, Beynon HL. Calcium metabolism in sarcoidosis and its clinical implications. Rheumatology (Oxford) 2000;39:707–713.
[10] Saidenberg-Kermanac’h N, Semerano L, Nunes H et al. Bone fragility in sarcoidosis and relationships with calcium metabolism disorders: a cross sectional study on 142 patients. Arthritis Res Ther 2014;16:R78.
[11] Baughman RP & Lower EE. Goldilocks, vitamin D and sarcoidosis. Arthritis Res Ther 2014;16:111.
[12] Joseph FG & Scolding NJ. Sarcoidosis of the nervous system. Pract Neurol 2007;7:234–244.
[13] Iannuzzi MC, Rybicki BA & Teirstein AS. Sarcoidosis. N Engl J Med 2007;357:2153–2165.
[14] Culver DA. Sarcoidosis. Immunol Allergy Clin North Am 2012;32:487–511.
[15] Nagai S, Handa T, Ito Y et al. Outcome of sarcoidosis. Clin Chest Med 2008;29:565–574.
[16] Scadding JG. Prognosis of intrathoracic sarcoidosis in England. A review of 136 cases after five years’ observation. Br Med J 1961;2:1165–1172.
[17] Joint statement on the American Thoracic Society (ATS), European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG). Statement on Sarcoidosis. Am J Respir Crit Care Med 1999;160:737–755.
[18] Cremers JP, Drent M, Bast A et al. Multinational evidence-based World Association of Sarcoidosis and Other Granulomatous Disorders recommendations for the use of methotrexate in sarcoidosis: integrating systematic literature research and expert opinion of sarcoidologists worldwide. Curr Opin Pulm Med 2013;19(5):546–561
[19] Drent M, Cremers JP, Jansen TL et al. Practical eminence and experience-based recommendations for use of TNF-α inhibitors in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:91–107.