Gombart, Sigrid / Science Photo Library
In 2013, the journal Science announced cancer immunotherapy as its “breakthrough of the year”. Since then, many have been asking whether the paradigm-shifting approach is the ‘answer to cancer’, or whether it will benefit only a minority of patients.
For attendees of the 2015 American Society of Clinical Oncology meeting in Chicago, Illinois, one of the largest annual gatherings of clinical cancer researchers in the world, it was impossible to ignore the buzz surrounding immunotherapy. The excitement centred around trials of ‘immune checkpoint inhibitors’ — therapies that switch off cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), which dampen the body’s immune response.
“The checkpoint inhibitors brought immunotherapy to the forefront of cancer therapy,” says Jay Berzofsky, an immunologist at the US National Cancer Institute (NCI) in Bethesda, Maryland. And Mark Schwartz, chief executive of Galena Biopharma, a Portland, Oregon-based company focusing on oncology, agrees. “The checkpoint inhibitors have validated the basic concept that unleashing T cells against cancer can be very effective,” he says.
The checkpoint inhibitors have validated the basic concept that unleashing T cells against cancer can be very effective
By removing the brakes from cytotoxic T lymphocytes — the subset of white blood cells that attack and kill virally infected or cancerous cells — checkpoint inhibitors allow them to expand and attack tumour cells. This often leads to long-lasting, durable responses in metastatic disease uncommonly seen with traditional anticancer therapy. And the numbers of patients who respond to checkpoint inhibitors are higher than with past immunotherapy approaches.
Often lost among the headlines, however, is the fact that the vast majority of cancer patients still do not benefit. “When checkpoint inhibition works, it really works, and when it doesn’t, it doesn’t,” says James Hodge, a cancer immunologist at the NCI, who estimates that only about 20% of all non-melanoma cancer patients respond to the therapies (the response rate in melanoma is particularly high).
Many cancer immunologists now think that in order for checkpoint inhibitors to work, the body must already be mounting an immune response, with T cells naturally infiltrating the tumours. One of the reasons melanoma is more sensitive to this type of therapy could be because it has a high mutational load, which may make it more of a target for the immune system. “If you are going to try to take the brakes off of T cells, there must be T cells there to take the brakes off of,” adds Hodge.

Source: US National Cancer Institute
James Hodge, a cancer immunologist at the US National Cancer Institute, says cancer vaccines are still a footnote in cancer immunotherapy
Stimulating an immune response
By killing cancer cells and releasing tumour proteins into the blood that can generate a T-cell response, some traditional anticancer agents may help stimulate the immune system. However, many cancer immunologists have long believed that the most effective way of kickstarting the immune response will be through therapeutic cancer vaccines, which directly prime T cells with these tumour proteins, or antigens.
Therapeutic cancer vaccines can be designed in a number of ways. Tumour antigens can be directly loaded on to the immune system’s antigen-presenting cells — dendritic cells — outside the body and then reintroduced. Or they can be administered in combination with viruses, bacteria or other substances that stimulate the immune system (adjuvants) to get dendritic cells to pick them up in vivo. Either way, the dendritic cells then travel to the lymph nodes to prime and activate T cells, so they can hunt down cancer cells expressing the tumour antigens.
“Many scientists are starting to wonder whether vaccine priming could increase the numbers of patients who would respond to checkpoint inhibitors,” says Mary Disis, a cancer immunologist at the University of Washington School of Medicine in Seattle. And John Sampson, a neurosurgeon at Duke University School of Medicine in Durham, North Carolina, predicts that: “checkpoint inhibitors will help herald a resurgence of therapeutic cancer vaccines.”
Many scientists are starting to wonder whether vaccine priming could increase the numbers of patients who would respond to checkpoint inhibitors
The potential of this combination has not been lost on pharmaceutical company Bristol-Myers Squibb (BMS), headquartered in New York, which has been at the forefront of developing immune checkpoint inhibitors. In March 2015, BMS signed an agreement worth up to US$975m with Danish biotech company Bavarian Nordic, based in Kvistgaard, giving it the exclusive option to license and commercialise Bavarian Nordic’s therapeutic prostate cancer vaccine PROSTVAC (PSA-TRICOM). The vaccine is in a 1,300-patient phase III trial in men with metastatic castration-resistant prostate cancer (CRPC)[1]
, and BMS is planning to test it in combination with its anti-CTLA-4 therapy ipilimumab (Yervoy).
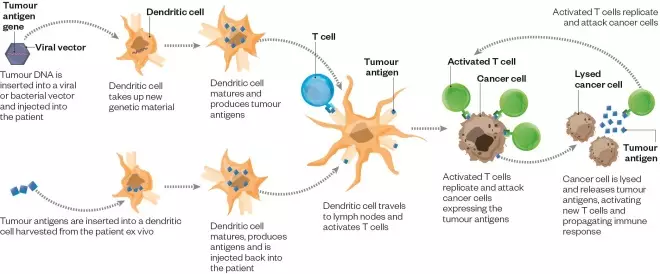
How therapeutic cancer vaccines work
Therapeutic cancer vaccines can be designed in a number of ways. For example, tumour antigens can be administered in combination with a virus or bacteria, which stimulates the immune system’s antigen-presenting cells — dendritic cells — to pick them up in vivo.
A ‘resurgence’ of therapeutic cancer vaccines is needed, as “cancer vaccines are still a footnote in cancer immunotherapy,” says Hodge, who has been involved in PROSTVAC’s development. After hundreds of clinical studies over the past couple of decades, just one such vaccine — Dendreon’s dendritic cell vaccine Provenge (sipuleucel-T) — has managed to secure US Food and Drug Administration (FDA) approval, in 2010, also to treat metastatic CRPC. And while many cancer immunologists feel that Provenge is an important development for the field, they are hoping that new vaccines will yield more than the four months median overall survival benefit it demonstrated in clinical trials.
Releasing the brakes
Among the most commonly cited reasons for the numerous failures of experimental therapeutic cancer vaccines is that most have been tested in metastatic disease. At this late stage, most tumours have developed a number of methods for keeping the immune system at bay, including taking advantage of the natural mechanisms that keep it from attacking host cells, such as T-cell immune checkpoints. “Vaccines have not worked in part because of immune checkpoints,” says Sampson. “If the brakes are released, they may be more effective.”
Some clinical studies combining vaccines and checkpoint inhibitors have already reported promising early results. In a randomised phase II trial of PROSTVAC, median overall survival was 25 months with the vaccine, 8.5 months longer than with the control[2]
. In a phase I combination trial with ipilimumab, it was 37 months for patients receiving the highest dose of ipilimumab.
Similarly, Amgen, based in Thousand Oaks, California, is developing an oncolytic viral immunotherapy called T-VEC (talimogene laherparepvec) that is nearing FDA approval for advanced melanoma. T-VEC is injected directly into tumours, where it replicates and finally ruptures tumour cells, releasing tumour antigens. In a phase III trial, it led to a durable response lasting at least 6 months in 16% of patients[3]
. This was improved to 44% of patients when used in combination with ipilimumab in a single-arm phase Ib/II study. “This synergistic effect is remarkable, we will see if it holds up,” says Howard Kaufman, a tumour immunologist at Rutgers Cancer Institute of New Jersey in New Brunswick, who has been involved in T-VEC’s development.
Measuring response
In addition to testing combination therapies, researchers are also attempting to develop improved vaccines that elicit more potent immune responses through better choices of antigens, adjuvants, and methods of delivery. “Things are working better than they ever have, but there is still a lot of room for improvement,” admits Hodge.
Things are working better than they ever have, but there is still a lot of room for improvement
One problem with trying to improve cancer vaccines is that it is difficult to measure how well they are working in early clinical trials. So far, no immune biomarker has been validated as a surrogate for an improvement in overall survival in a large phase III cancer vaccine trial.
“There are different ways of measuring T-cell responses — their quantity, their quality, their breadth, their avidity. Which of these translates into antitumour effect in vivo is harder to figure out,” says Nina Bhardwaj, a cancer immunologist at Mount Sinai School of Medicine in New York. According to Berzofsky, a T-cell’s functional avidity, or the strength with which it binds antigens, reflected in its sensitivity to low levels of antigen, is most important. “One high-avidity T cell is worth many low-avidity T cells, as higher avidity T cells can respond to lower concentrations of antigens often found on tumour cells that could not even be detected by low-avidity T cells.”
One way of improving T-cell avidity is to deliver costimulatory molecules — signals generally expressed by antigen-presenting cells to get T cells proliferating — along with the vaccine antigens. PROSTVAC and another vaccine, PANVAC, include genes for three such molecules — B7.1, ICAM-1 and LFA-3, together called TRICOM — in their poxvirus vaccine vectors.
But for Jeffrey Weber, a cancer immunologist at Moffitt Cancer Center in Tampa, Florida, most vaccines have not evoked sufficient quantities of antigen-specific T cells. “Show me a cancer vaccine that routinely yields 5–10% of circulating T cells that are antigen-specific, as in an acute viral infection, and I’ll be impressed,” he says. Therapeutic cancer vaccines have more typically yielded closer to 0.1% of antigen-specific T cells, he adds.
According to Hodge, this is likely to be an underestimate, on account of the way current assays typically measure T-cell response. Still, he admits that cancer vaccines elicit less of an immune response than observed during a viral infection. While this level of response may not completely eliminate metastatic disease, “a win for now is increased overall survival and tumour stability,” he says.
Choice of antigens
The immune system responds much more vigorously to viral antigens because they appear naturally foreign to it. In contrast, many tumour-associated antigens appear as “self” — for example, some may appear in small amounts on healthy cells, which the immune system has learnt to ignore. “We’ve been barking up the wrong tree with respect to antigens,” says Weber.
He and many others are excited about a new class of antigens being investigated, so-called neoantigens, which are derived from mutated proteins specific to an individual patient’s tumour.
Recent advances in genome sequencing have enabled the first phase I clinical trials of personalised neoantigen vaccines. In a paper published online in Science in April 2015, which garnered significant attention, Beatriz Carreno from Washington University School of Medicine and colleagues, tested a personalised neoantigen vaccine in three melanoma patients whose tumours had been surgically resected, but who were at significant risk of recurrence[4]
.
The researchers used dendritic cells to deliver neoantigens derived from the individual tumours. They found that each patient had a pre-existing T-cell response to one mutated neoantigen included in the vaccine, and the vaccine was able to trigger an immune response against two additional neoantigens. “Before vaccination, very few clones of T cells could be detected for each of the antigens. After vaccination, there was an explosion of antigen-specific T-cell clones,” says Carreno. Between 0.1% and 0.9% of T cells in the blood after vaccination were antigen-specific.
But dendritic cells may not be the best choice of delivery vehicle for tumour antigens, Weber says. “Dendritic cells are fragile, not enough of them typically survive after injection into patients to prime enough T cells,” he adds. At the Dana-Farber Cancer Institute in Boston, Massachusetts, oncologist Catherine Wu and colleagues are instead developing personalised neoantigen vaccines using a pool of long peptides as antigens, along with an adjuvant to stimulate an immune response in vivo.
Both Carreno and Wu say that tumour types with the highest mutational load are the best types to test neoantigen vaccines in initially, as it is more likely they will generate antigens that will be presented to T-cells.
Another limitation of personalised vaccines is that they currently take around four months to make — too long for rapidly progressing metastatic disease. For this approach to be used to treat advanced disease, the process will need to be faster.
While most neoantigens appear to be passenger mutations — that is, they occur in tumour cells but are not driving tumour growth — driver mutations that are commonly shared between patients with a specific tumour type can also be used as vaccine antigens. Hampton, New Jersey-based Celldex Therapeutics’s peptide vaccine, RINTEGA (rindopepimut)[5]
, is based on such a mutation — epidermal growth factor receptor variant III (EGFRvIII), which occurs in approximately 30% of glioblastoma patients. In a phase II study in recurrent glioblastoma, the vaccine improved median overall survival by a couple of months, leading to a phase III trial in newly diagnosed glioblastoma.
However, in another trial, Sampson, whose lab developed the vaccine, and colleagues found that at recurrence, more than 80% of patients had lost EGFRvIII expression in their tumours. “Vaccines that target only one antigen may not target all tumours or all cells comprising a tumour, and may therefore select for the survival and proliferation of those cells that do not express the targeted antigen,” they write[6]
.
Many point to this danger of antigen loss as a reason to develop vaccines that deliver multiple antigens. Wu and her team are planning to include as many as 20 antigens in their personalised neoantigen vaccines to address the genetic heterogeneity of the tumour. Use of multiple antigens, and multiple epitopes (sequences that are recognisable to the immune system) within a single antigen, also evokes “a greater quantity of T cells that have activity against the tumour”, says Disis.
However, even single-antigen vaccines, like PROSTVAC, have been shown to induce immunity against other important, unknown, cancer antigens in the tumour. This ‘epitope spreading’ or ‘antigen cascade’ effect seems to occur when T cells that infiltrate the tumour encounter and are primed with new antigens, the extent of which may predict clinical benefit[7]
. “If we get the fire started, the immune response can propagate on its own,” says Hodge. However, he adds that the fire may get going faster if multiple, relevant antigens are included in the vaccine upfront to “recapitulate the antigen cascade from the start”.
The most common antigens included in cancer vaccines have been non-mutated ‘self’ antigens that are overexpressed in tumours compared with normal tissue, such as prostate-specific antigen; only expressed normally during foetal development; expressed in altered forms in cancer; or are tissue-specific.
These antigens have the advantage of being commonly shared between patients with a specific tumour type, meaning that it is unnecessary to develop personalised vaccines. “Trying to treat every patient with a personalised vaccine is a daunting challenge,” says Hodge.
Trying to treat every patient with a personalised vaccine is a daunting challenge
It is not yet clear whether mutated or overexpressed tumour antigens lead to the strongest immune response.
Adjuvant setting
Although the immune response elicited by cancer vaccine monotherapy so far may not be sufficient to eliminate bulky, metastatic disease, it might more effectively eliminate tiny, undetectable micrometastases to reduce the rate of disease recurrence following surgical removal of a localised tumour.
In the adjuvant setting, cancer vaccines could lead to an antigen-specific memory T-cell response that lasts for an extended period of time, potentially a lifetime. And the cancer may also be less likely to develop resistance than with traditional therapy, as long as the vaccine can generate a broad-based immune response.
“The setting is one of the most critical components of making a vaccine work,” says Galena’s Schwartz. The company is testing its vaccine, NeuVax (nelipepimut-S), which includes a peptide antigen derived from the HER2 protein, in the adjuvant setting in a phase III trial in breast cancer.
However, according to Olivera Finn, a cancer immunologist at the University of Pittsburgh, Pennsylvania, even the adjuvant setting might be too late for vaccine monotherapy. “The patient’s immune system is still left with the imprint of the primary tumour after it has been removed,” she says. She worries that the immunosuppressive mechanisms activated by the tumour remain in place.
The most appropriate role for cancer vaccines, Finn believes, is in the prevention of cancer. With her collaborators, she has launched a 120-patient, NCI-sponsored randomised phase II trial to test a vaccine designed to prevent the recurrence of colorectal adenomas — premalignant precursors of colorectal cancer[8]
. Finn turns to history to support her approach. “Other than rabies, there is no precedent for an effective therapeutic vaccine in infectious diseases, so this is an unrealistic expectation of cancer vaccines.”
References
[1] Bavarian Nordic. A randomized, double-blind, phase 3 efficacy trial of PROSTVAC-V/F +/- GM-CSF in men with asymptomatic or minimally symptomatic metastatic castrate-resistant prostate cancer (Prospect) . ClinicalTrials.gov identifier NCT01322490.
[2] PW, Schuetz TJ, Blumenstein BA et al. Overall survival analysis of a phase II randomized controlled trial of a poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. Journal of Clinical Oncology 2010;28:1099–1105.
[3] Andtbacka RHI, Kaufman HL, Collichio F et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. Journal of Clinical Oncology 2015.
[4] Carreno BM, Magrini V, Becker-Hapak M et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015;348:803–808.
[5] Celldex Therapeutics. Phase III study of Rindopepimut/GM-CSF in patients with newly diagnosed glioblastoma (ACT IV). ClinicalTrials.gov identifier NCT01480479.
[6] Sampson JH, Heimberger AB, Archer GE et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of Clinical Oncology 2010;28:4722–4729.
[7] Gulley JL, Madan RA, Tsang KY. Immune impact induced by PROSTVAC (PSA-TRICOM), a therapeutic vaccine for prostate cancer. Cancer Immunology Research 2014;2:133–141.
[8] University of Pittsburgh. Vaccine Therapy in Treating Patients with Newly Diagnosed Advanced Colon Polyps. ClinicalTrials.gov identifier NCT02134925.