SCIENCE PHOTO LIBRARY
After reading this article, you should be able to:
- Explain what an advanced therapy investigational medical product (ATIMP) is;
- Describe the typical process of using an ATIMP in a clinical trial;
- Describe the role of the pharmacist in ATIMP trials;
- Understand the main issues and considerations of using an out-of-specification ATIMP.
Introduction
Advanced therapy medicinal products (ATMPs) offer a novel method of treating or preventing disease. ATMPs can be classified, depending on their starting material, as gene therapy, somatic cell therapy or tissue-engineered products[1]. They can also incorporate medical devices, in which case they are known as combined ATMPs. For more information on the clinical, institutional, regulatory, safety and economic considerations of advanced therapy medicinal products, see ‘Advanced therapy medicinal products: a comprehensive overview for pharmacy professionals‘.
Prior to regulatory approval, ATMPs are called advanced therapy investigational medicinal products (ATIMPs) and have particular logistical, technical and clinical considerations when assessing their suitability and appropriateness for use. This is owing to their novelty and complex nature of biological origin and technical specificity, which poses new challenges for the NHS.
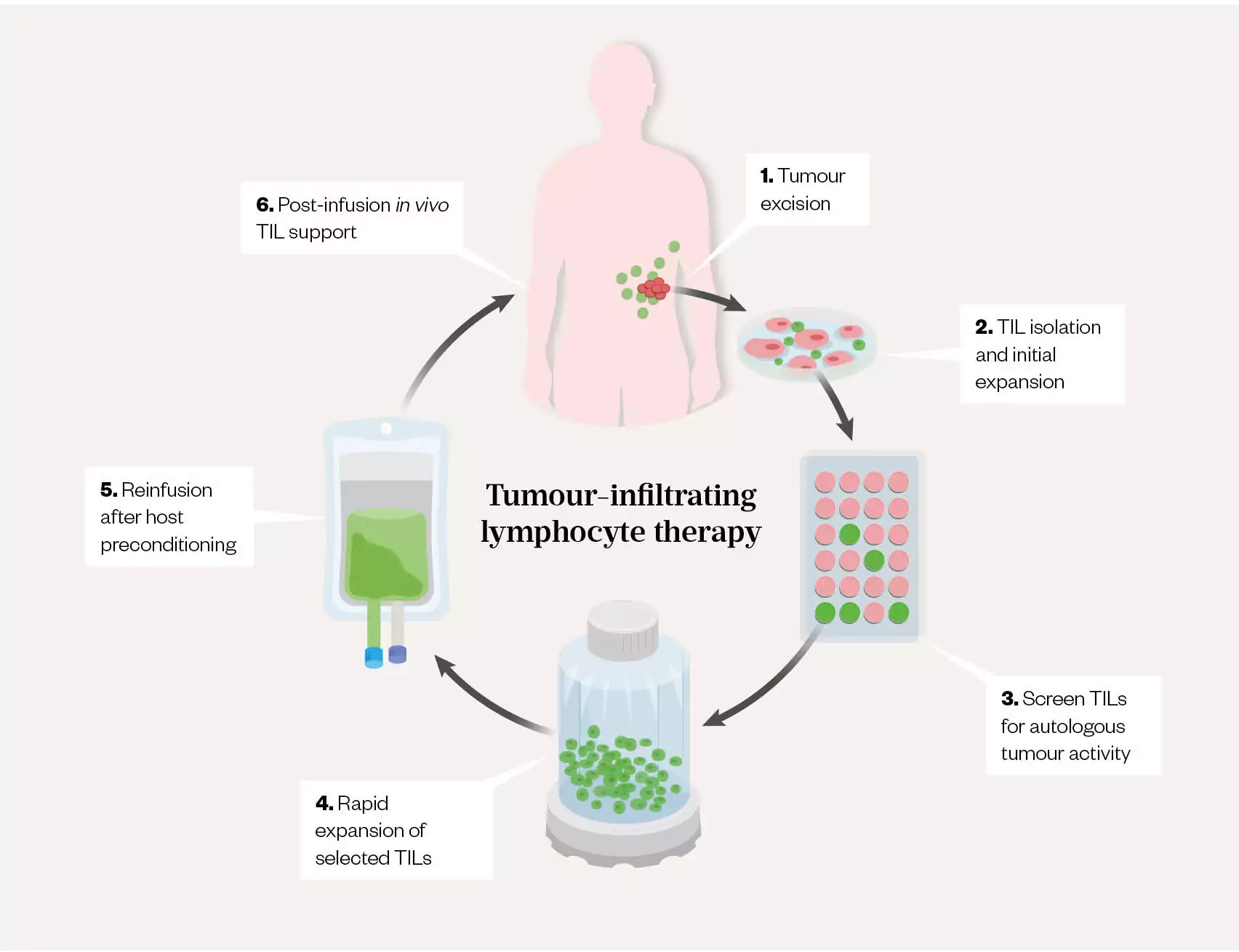
Tumour-infiltrating lymphocyte (TIL) therapy is an example of an autologous cell-based ATMP, whereby the TILs are harvested from the tumour site and expanded (increased in number) ex vivo (see Figure 1)[2]. The tumour-specific and patient-specific mutations (neoantigens) are identified by biopsy and tumour sequencing. The patient’s T-lymphocytes from the tumour are then assessed to identify and isolate lymphocytes with antigens that are specific to these tumour mutations; however, these antigens are silenced in the tumour microenvironment. The isolated T-lymphocytes are activated and expanded ex vivo (see Figure 2) and comprise predominantly T-cells with multiple T-cell-receptor clones, which will attach to the neoantigens. Therefore, when the TILs are re-infused back into the patient, the TILs now have an enhanced ability to recognise and target the neoantigens, resulting in tumour-specific cytolysis through a patient-tailored approach[2–5]. This adoptive cell therapy (i.e. whereby endogenous cells are isolated, selected, expanded and re-infused) is currently being investigated in treating solid tumours[2,5].
This case-based learning article discusses the practical considerations dealt with when an investigative site was faced with a TIL ATIMP that did not comply with the product release specification and was therefore out of specification. It is based on a real case study that surfaced a need for novel decision-making in a fast developing therapeutic area. As the field of advanced therapies grows, the potential for administration of an out-of-specification ATIMP will increase. It is particularly important for hospital pharmacists to be aware of the considerations and governance requirements necessary when assessing and approving these medicinal products for administration.
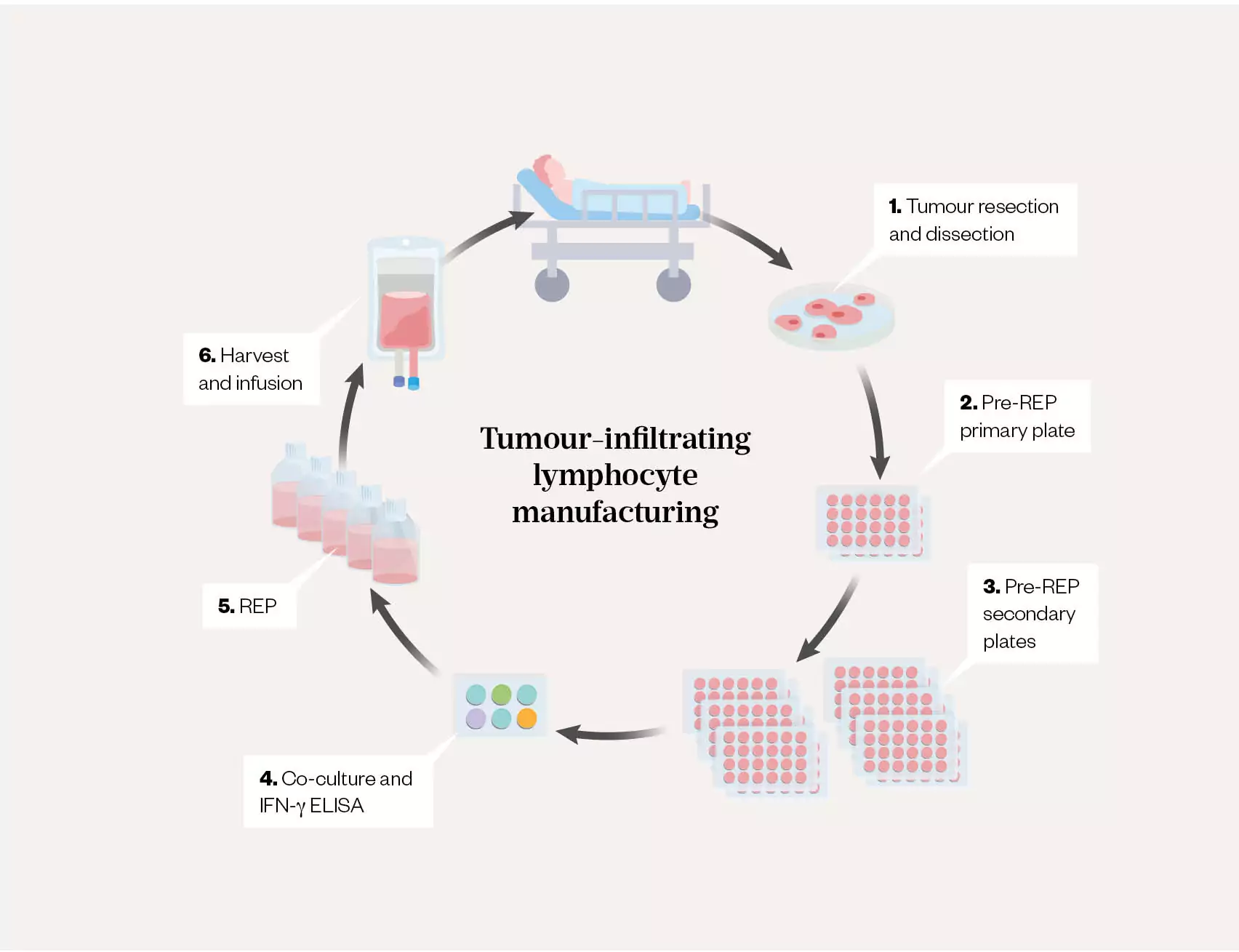
REP: rapid expansion protocol; IFN-γ = interferon-γ; ELISA: enzyme-linked immunosorbent assay
The role of the pharmacist in ATIMP trials
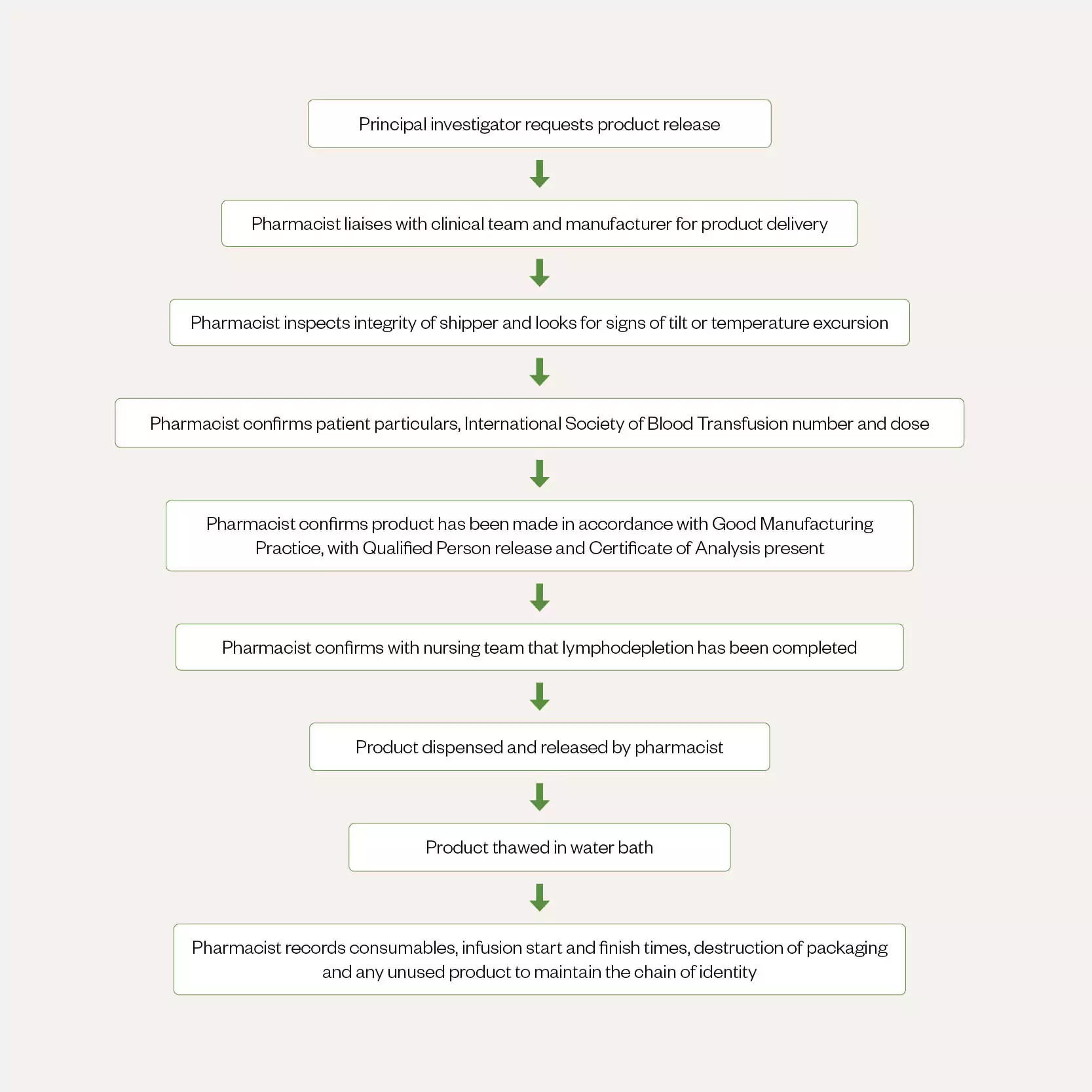
In a phase 1 clinical study at University Hospital Southampton (UHS) NHS Foundation Trust, the ATMP pharmacist is involved from the point of receipt of the manufactured medicinal product. ATMPs require specialist handling, as they contain living cells. Pharmacy must have oversight of these products at all stages: receipt, storage, handling, administration and destruction (see Figure 3).
The ATMP pharmacist liaises with the clinical team and the manufacturer to receive the product on the ward. The cell-therapy product is delivered in a dry nitrogen ‘shipper’, which must be inspected for signs of damage or tampering, tilting, and any temperature excursion before receipt. The shipper has cable ties specific to the patient, displays a tilt-watch and has an electronic temperature logger attached to show any temperature deviation in transit. The product itself must be inspected against the order details and prescription to ensure that it is for the correct patient. It should be noted that the dosing format of cell-based products differs from that of other medicinal products. Cell-based products, such as chimeric antigen T-cell (CAR-T) products, have a target dose range with exponents in terms of ‘x10x’. The trial protocol or summary of product characteristics will state the target volume of cells for administration.
The ATMP pharmacist should also confirm that the product has been manufactured in accordance with the correct specification (presence of Qualified Person [QP] certification and Certificate of Analysis [CofA]). Working with the clinical trial/bone marrow-transplant nurse, the pharmacist should confirm that lymphodepletion has taken place and proceed to dispensing and thawing the product using a water bath. Owing to the short shelf-life of these products, thawing must be completed outside of the patient’s treatment room. Following cell thaw and confirmation that the cryobag is intact, the cells are administered by the nurse. The pharmacist’s role is to record all consumables used, infusion start and finish times, destruction of packaging and any unused product. This maintains the chain of identity.
Case presentation
A patient aged 73 years was diagnosed with non-small cell adenocarcinoma of the lung (NSCLC) with malignant neoplasm of pleura in 2020. They had a history of smoking, having smoked one pack of cigarettes per week when aged 15–21 years, with the occasional cigar since. They were drinking around 18 pints of beer per week and working full-time in a warehouse.
Upon diagnosis, immunotherapy with pembrolizumab was started promptly. Following disease progression in July 2021, second-line treatment of chemotherapy with carboplatin and pemetrexed was commenced. However, following tinnitus and declining renal function, the treatment was reviewed in early 2022. Options available at this point were: restarting chemotherapy with docetaxel and nintedanib (predicted response rate of 10%); targeted treatments (should molecular testing yield any positive results); or supportive care or trial treatments. From tumour profiling and genomic sequencing, no therapies with clinical specificity to the patient’s tumour type were identified. Following discussion with the patient’s clinical team and wider family, the patient decided to enrol in a Phase 1 clinical trial investigating the use of personalised TIL therapy in solid tumours.
In March 2022, the patient underwent tissue biopsy from the primary site of their lung cancer to manufacture the clonal neoantigens required for the TIL therapy. The procurement and cell harvesting took place onsite at UHS. These samples were sent to the manufacturer and the ATIMP manufacturing process took 10–12 weeks.
During the manufacturing process, only 0.4 x 107 viable reactive CD3+ cells were produced, instead of the target dose of 1 x 107 to 1 x109 CD3+ cells. Therefore, this product was classed as an out-of-specification ATIMP. The sequence of events was as follows:
- The manufacturer/sponsor alerted the principal investigator to this out-of-specification product and enquired whether the principal investigator still wished to treat;
- The principal investigator (who was the patient’s treating consultant) undertook a clinical risk assessment and discussed this out-of-specification product with the patient. The principal investigator discussed the risks and benefits with peers experienced in phase 1 trials and ATMPs. From this, the principal investigator and patient made a joint decision that it was in the patient’s best interest to receive the out-of-specification ATIMP. The principal investigator therefore formally requested for the sponsor to release the product to site.
- One week prior to cell return, the principal investigator and the ATMP pharmacist received the ‘Sponsor release and authorisation to ship form’, which stated that the ATIMP dose was out of specification (as expected) and that it would not be QP-certified (this was not expected). At this point, the patient had already commenced lymphodepletion.
- Upon discussion with the sponsor, the QP confirmed that the cells were manufactured and released with quality assurance (QA) verification with compliance to good manufacturing practice (GMP), and that a CofA was available.
- Because of this unprecedented scenario, the ATMP pharmacist consulted with the UHS aseptic services pharmacists, pharmacy trials pharmacist and bone marrow transplant QA team, and sought advice from the Specialist Pharmacy Service Regional QA specialist and chair of the Pan UK Pharmacy Working Group for ATMPs. Published guidance was also checked[6].
- The principal investigator, ATMP Oversight Committee chairs, chief pharmacist, clinical trials lead pharmacist and ATIMP pharmacist discussed the out-of-specification ATIMP use. The multidisciplinary team discussed the risks of the out-of-specification product, potential risks to the patient of both treating and not treating, and ethical and financial considerations of its administration. Having assessed the clinical need, in conjunction with the critical nature of the treatment and the potential risks of the out-of-specification ATIMP, it was decided that treating with the out-of-specification ATIMP was in the patient’s best interest. The summary of this risk assessment is outlined in the Table.
A plan was then made to communicate this event with the Medicines and Healthcare products Regulatory Agency by the sponsor. This decision was documented on the patient’s electronic care record. The UHS team modified its current ‘Free-of-charge/compassionate use medicine approval form’. This was to guide and document these discussions and assessments and was reviewed by the chief pharmacist and ATMP Oversight Committee co-chair.
Product release documentation
A QP is responsible for the final certification of an investigational medicinal product (IMP), ensuring compliance with any specifications for GMP as per the Investigational Medicinal Product Dossier (IMPD), with the appropriate pharmaceutical quality system in place (i.e. Annex 13)[7]. Ordinarily, the UHS team would expect EU/GB QP certification for any investigational medicinal product received on site. When an investigative site receives a product, the sponsor should provide either a QP certificate or an equivalent sponsor statement.
Previously, when an IMP (not advanced therapy) has been released from the manufacturer without a QP certification to UHS, the product has been supplied and administered as a ‘special’ for compassionate use and on an off-trial basis. This option was discussed with the sponsor and with the chair of the Pan UK Pharmacy Working Group for ATMPs. The advice provided was that, owing to the nature of ATMPs and the extent of deviation from specification, the individual patient must be considered. ATIMPs are of biological origin and intrinsically unique, and therefore have a greater potential for variation[6]. This may increase the chance of them being out of specification. Therefore, there may be cases where it is in the best interest of the patient that an out-of-specification ATIMP is administered, depending on the degree of non-compliance with the original specification and contributing risk factors. A risk–benefit assessment was advised for this individual case.
In this scenario, the sponsor was unable to provide a QP certificate, as the product did not comply with the specification in the IMPD. It was confirmed that the out-of-specification parameter was that the quantity of cells produced was less than that stated on the IMPD, but that other quality parameters were unaffected (such as sterility and endotoxin levels). The QP provided written verification that the product was made to GMP standards. This verification was also documented on the CofA provided. The sponsor provided a risk assessment, comprising clinical and technical considerations, to guide the principal investigator in making their clinical decision. A Deviation and Exceptional Report Notification was also put in place, as this was a significant shift from usual trust practice when receiving the ATIMP stock for clinical trial use.
Responsibility
The manufacturer/sponsor did not waive all responsibility for the product, as they provided a written risk assessment and their QP provided details of the product results achieved against the expected specification outlined in the IMPD. They also verified that this was in accordance with GMP standards.
However, because of the absence of a QP certificate and the wording of the out-of-specification ATIMP product request form, the responsibility of dosing shifted from the sponsor to the principal investigator and chief pharmacist. To ensure the correct governance procedures were in place and support the principal investigator in terms of responsibility and decision-making process, the out-of-specification ATIMP administration was formally discussed and approved at a trust level.
Patient considerations
The patient had received no active treatment since the beginning of 2022 and had significant disease progression, with no other treatment options. Immediate action was therefore essential owing to the patient’s condition. It was unlikely that the patient would be fit enough to undergo another biopsy and wait 10–12 more weeks for cell production. Furthermore, even if this was possible, it was unlikely to yield sufficient cells for the target dose. The patient re-consented to receive this cellular therapy, with full knowledge that the product would be out of specification.
The patient had commenced lymphodepletion, in preparation of cell return, and was due day three of their lymphodepletion on the day that the ATIMP committee reviewed their case; therefore, the need to treat with the autologous cells was very urgent. This was a phase 1 study investigating safety, tolerability and clinical efficacy; therefore, there were already a significant number of unknown factors and risks to the patient.
Trial considerations
The QP’s verification that the ATIMP was manufactured in accordance with GMP was vital in the decision to administer treatment to the patient. The impact of the out-of-specification product on the investigative treatment had to be assessed, with the following questions posed:
- Would the patient be treated on the trial?
This was different to previous non-ATIMP studies at UHS, whereby if the IMP was classed as out of specification or not as per IMPD, the patient would no longer be treated on the trial. - Could the patient stay on the trial after administration?
This was less urgent, but the answer would have ramifications for the wider multidisciplinary team regarding follow-up monitoring and review.
These two questions were discussed at the ATMP Oversight Committee meeting as part of the out-of-specification product approval process. Confirmation that the patient was still to be treated on-trial was sought from the sponsor. It was confirmed that the MHRA would be notified (time did not permit notification pre-dosing), and that follow-up was to be done on-study as per protocol. Use of the patient data in trial results would be discussed and determined at a later point by the sponsor.
The wider implications of treating off-trial, including trust capacity and financial considerations, were discussed by the ATMP Oversight Committee. The trial schedule entailed only a single dose of ATIMP with no further treatment. However, had this been a different study, the impact on future dosing (where appropriate), and who would cover these costs, would also need to be considered. These include the potential for further ATIMP dosing, hospital admissions, adverse event and side effect management, chemotherapy/immunotherapy add-on treatment, monitoring requirements and necessary scans.
Conclusion
An ATIMP can be out of specification for several reasons and to various degrees of severity. The multidisciplinary team’s decision to administer an out-of-specification ATIMP should follow local governance and be reviewed and determined at a trust level. At UHS, the ATMP Oversight Committee met and made a decision, considering the patient’s condition, the sponsor’s risk assessment and the principal investigator’s clinical risk assessment.
This was the first time at UHS that an ATIMP, and in fact an IMP of any kind, that was not QP-certified was to be administered as part of a phase 1 trial. This case has highlighted the additional considerations faced when assessing out-of-specification IMPs in a clinical trial. After this case, the UHS team devised an internal standard operating procedure and an ‘out-of-specification AT(I)MP request form’, which prompts discussion and evaluation of each risk (safety, financial, ethical and clinical). It also highlighted the need to work collaboratively across the multidisciplinary team, involving the treating physician/principal investigator, ATMP Oversight Committee and pharmacy team. In the future, collaboration might also include a member of the Clinical Ethics Committee and the facility where the cells are manufactured.
The need to administer out-of-specification ATIMPs is likely to increase over time, as more ATIMPs are involved in clinical trials and become available on the market for licensed use. It is therefore essential for all clinical sites to proactively consider the Specialist Pharmacy Service guidance on this subject and put robust procedures in place for managing and approving out-of-specification ATMP/ATIMP use and to ensure pharmacy teams play a central role.
Acknowledgements
The authors would like to thank the patient for providing their consent to share this case study, the clinical trial sponsor for their support in the publication of this article and Anne Black, chair of the Pan UK Pharmacy Working Group for ATMPs.
Conflict of interest statement
The authors declare no support from any organisation for the submitted work, no financial relationships with any organisations that might have an interest in the submitted work and no other relationships or activities that could appear to have influenced the submitted work.
- This article was amended on 22 May 2023
- 1Regulation (EC) No 1394/2007 of the European Parliament and of the Council. EU Council. 2007.https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:en:PDF#:~:text=The%20scope%20of%20this%20Regulation,of%20the%20Community%20pharmaceutical%20legisla (accessed Apr 2023).
- 2Qin SS, Melucci AD, Chacon AC, et al. Adoptive T Cell Therapy for Solid Tumors: Pathway to Personalized Standard of Care. Cells. 2021;10:808. doi:10.3390/cells10040808
- 3Hopewell EL, Cox C, Pilon-Thomas S, et al. Tumor-infiltrating lymphocytes: Streamlining a complex manufacturing process. Cytotherapy. 2019;21:307–14. doi:10.1016/j.jcyt.2018.11.004
- 4van den Berg JH, Heemskerk B, van Rooij N, et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: boosting of neoantigen-specific T cell reactivity and long-term follow-up. J Immunother Cancer. 2020;8:e000848. doi:10.1136/jitc-2020-000848
- 5Wang S, Sun J, Chen K, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021;19. doi:10.1186/s12916-021-02006-4
- 6Out of Specification Advanced Therapy Medicinal Products – Guidance for Healthcare Organisations. Specialist Pharmacy Service. 2020.https://www.sps.nhs.uk/articles/out-of-specification-advanced-therapy-medicinal-products-guidance-for-healthcare-organisations/ (accessed Apr 2023).
- 7EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines. European Commission. 2009.https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en (accessed Apr 2023).