

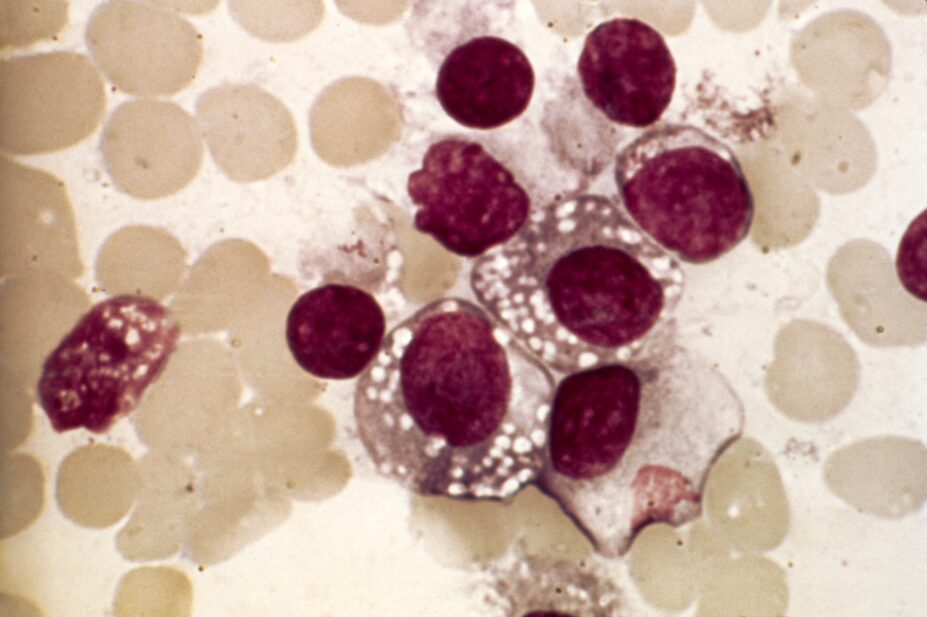
PR. J. BERNARD/CNRI/SCIENCE PHOTO LIBRARY
After reading this article, you should be able to:
- Describe the indications for treatment of Waldenstrom’s macroglobulinaemia;
- Understand the molecular characteristics and determinants of response to BTK-directed therapy;
- Recognise the common toxicities of treatment.
Introduction
Waldenstrom’s macroglobulinaemia (WM) is a rare type of non-hodgkin lymphoma, characterised by the presence of a monoclonal IgM protein[1,2]. Approximately 350 people are diagnosed with WM in the UK each year[3]. It is more common in men than it is in women, and it is much more common among people of white ethnicity than people of black ethnicity[4]. In approximately 20% of cases, there is a familial predisposition[5]. Approximately 1.5–2.0% of healthy individuals with an IgM-MGUS (monoclonal gammopathy of undetermined significance) progress to WM each year[6]. As with other chronic B-cell malignancies, WM tends to affect older people, with an average age at diagnosis of 70 years[7]. It is incurable, with median survival of 10–12 years; however, many patients die from causes unrelated to the disease[7].
Symptoms of disease can arise from the physico-chemical and immunological effects of the monoclonal IgM protein (paraprotein) or from infiltration of the bone marrow and lymphoid organs; in the absence of symptoms, patients can be monitored with active surveillance[8].
Although traditionally managed with chemo-immunotherapy, targeted treatments, such as Bruton’s tyrosine kinase inhibitors (BTK-Is), are highly effective and are becoming the mainstay of treatment for WM.
This article will outline the symptoms of WM and how it is diagnosed, and will detail how the underlying pathophysiology can guide management of the condition, as well as provide an overview of the current available treatment options.
Cancer learning ‘hub’
Pharmacists are playing an increasingly important role in supporting patients with cancer, working within multidisciplinary teams and improving outcomes.
However, in a rapidly evolving field with numbers of new cancer medicines is increasing and the potential for adverse effects, it is now more important than ever for pharmacists to have a solid understanding of the principles of cancer biology, its diagnosis and approaches to treatment and prevention.
This new collection of cancer content, brought to you in partnership with BeOne Medicines, provides access to educational resources that support professional development for improved patient
Pathophysiology
WM arises from malignant transformation of lymphoplasmacytic B cells. The origin of the malignant clone is thought to be a B cell arrested after somatic hypermutation in the germinal centres and before terminal differentiation to plasma cells. These are B cells that have not undergone class-switch recombination and secrete polyclonal IgM as an early phase immune response to infection. Malignant transformation leads to increased proliferation and survival of these cells, leading to expansion of the clone and secretion of a monoclonal IgM paraprotein[1].
The Pharmaceutical Journal
Chromosome 6q deletions have been observed in up to half of patients with WM[9]. The presence of 6q deletions has been suggested to distinguish patients with WM from those with IgM-MGUS. Other chromosome abnormalities detected by cytogenetic or fluorescent in situ hybridization analyses include deletions in 13q14, 17p, and 11q and trisomy 4, 12 and 18[10].
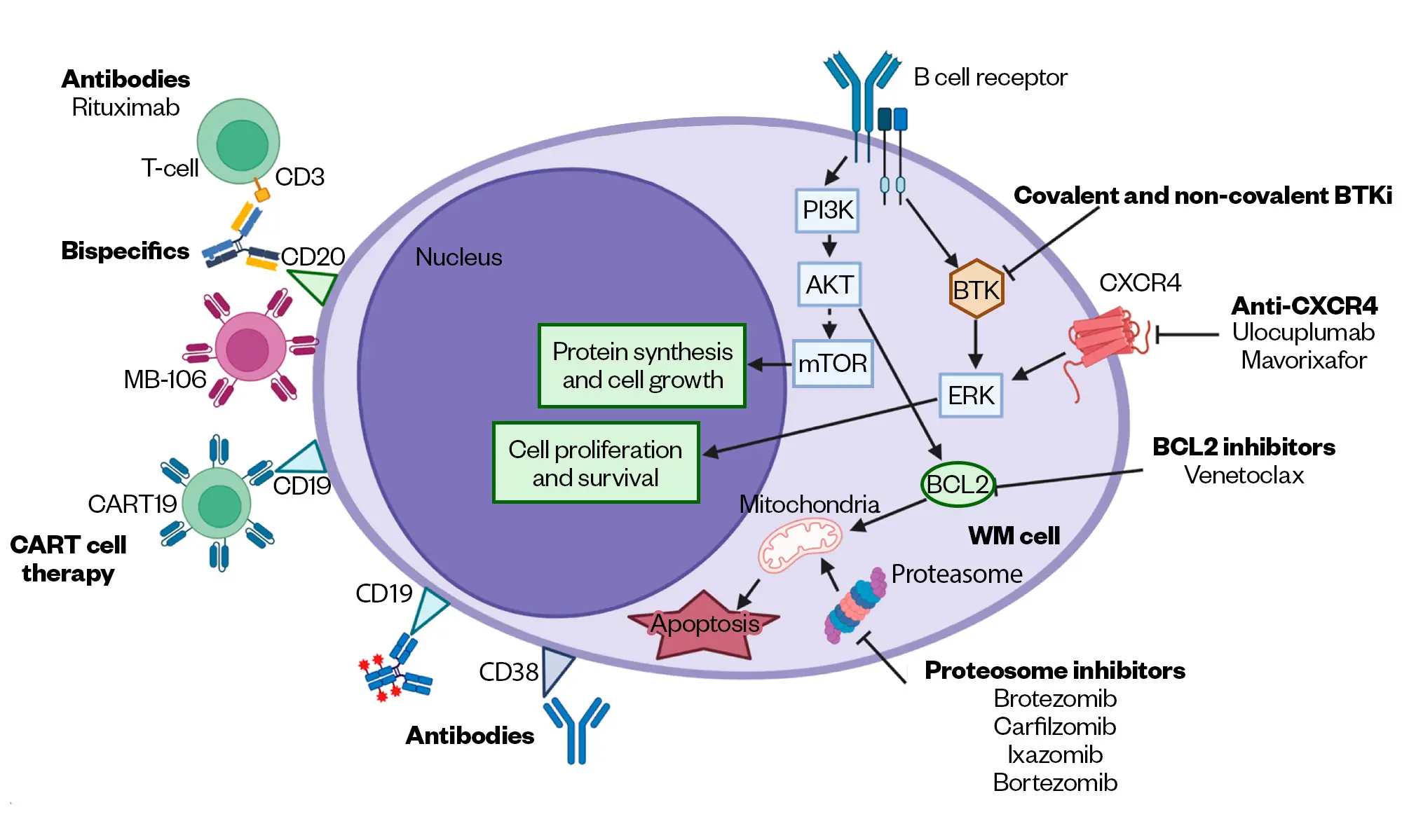
MYD88 and CXCR4 mutations affect WM disease presentation and treatment outcome.
In 95–97% cases, there is a single-activating somatic mutation in MYD88, leading to the substitution of leucine with proline (L265P), which triggers BTK and activates growth and survival signalling via NFκB11 (Figure 1)[11]. Patients with wild-type MYD88 show lower bone marrow disease burden and serum IgM levels but have an increased risk of death[12].
MYD88 mutations may be detected by allele-specific polymerase chain reaction (AS PCR) or by targeted next-generation sequencing (NGS). A study that screened 734 WM patients using both NGS and AS PCR demonstrated that there was a significant discordance with NGS, leading to false negative results in approximately 30% of patients; therefore, laboratories should specify the method used and its sensitivity[13].
Somatic-activating mutations in the C-terminal domain of CXCR4 are present in up to 40% of patients with WM and are nearly always observed in conjunction with MYD88 mutations in patients with WM[14]. These mutations are similar to germline mutations detected in patients with WHIM syndrome (warts, hypogammaglobulinemia, infections, and myelokathexis). In WM, somatic CXCR4 mutations result in impaired internalisation of the CXCR4 receptor, leading to constitutive activation of the CXCR4 pathway, protein kinase B (Akt) and extracellular signal-regulated kinase (ERK) activation and, subsequently, WM cell survival. Patients with CXCR4 mutations, particularly those with nonsense mutations, have higher bone marrow disease burden, higher serum IgM levels, are more likely to develop hyperviscosity and have an increased propensity to ibrutinib resistance compared with CXCR4 wild-type patients[15–17].
Histological transformation, primarily to diffuse large B-cell lymphoma, is a well recognised phenomenon in WM, with a ten-year incidence of approximately 2.4%[18].
Symptoms
WM represents a spectrum of disease, with many people having no symptoms at all. When symptoms arise, they may be related to the presence of the IgM paraprotein or infiltration of the bone marrow and tissue with clonal lymphocytes, or they may be constitutional in nature (e.g. fatigue, loss of appetite and weight loss)[19].
The IgM molecule is a pentameric structure, which at significant levels (>40g/L) may cause symptomatic hyperviscosity. Clinical features of hyperviscosity include headache, visual disturbance and mucosal bleeding. The IgM paraprotein can cause symptoms through its immunological effects; these include symptoms relating to the formation of cryoglobulins (proteins that precipitate from an individual’s serum or plasma at temperatures lower than 37°C) and include Raynaud’s phenomenon (peripheral circulatory effects), skin changes and neuropathy. IgM-related red cell agglutination can cause haemolytic anaemia and acrocyanosis, tissue deposition in the form of amyloid fibrils, fatigue, weight loss and organ dysfunction related to the heart, kidney, liver and peripheral sensory and autonomic nerves. Rarely, patients may develop a bleeding diathesis, including epistaxis or spontaneous haematoma, as a consequence of secondary von Willebrand disease. Iron deficiency anaemia may arise as a result of dysregulated iron homeostasis[19].
Infiltration of lymphoid tissue by clonal lymphocytes can cause discomfort related to lymphadenopathy and hepatosplenomegaly. Infiltration of the bone marrow can lead to underproduction of red blood cells, white blood cells and platelets, leading to tiredness, breathlessness, increased risk of infection and a predisposition to bleeding and bruising[19].
WM-related peripheral neuropathy is reported by 25% of newly diagnosed WM patients as their presenting symptom[20]. The clinical spectrum spans from distal paraesthesias (i.e. abnormal skin sensations in the extremities) and mild gait imbalance to more severe sensory ataxia (i.e. loss of coordination), with falls and a varying degree of both sensory and motor deficits in the extremities. WM-related peripheral neuropathy is most commonly associated with anti-myelin associated glycoprotein (MAG) and anti-ganglioside antibodies, but may also be caused by amyloid light-chain (AL) amyloidosis or the presence of cryoglobulins[20].
Rarely (affecting 1% patients during the disease course), lymphoplasmacytic lymphoma (LPL) can infiltrate the central nervous system (Bing–Neel Syndrome), leading to clinical presentation with a spectrum of neurological symptoms, from headache/visual disturbance to seizures and cognitive decline to cranial neuropathies and sensory disturbance according to the localisation of disease[21]. The diagnosis is made following examination of imaging and cerebrospinal fluid analysis.
Diagnosis can be challenging owing to the varied nature of WM presentation. Click play below to listen to Harriet Scorer, a patient trustee of WM UK, to hear about her experience of being diagnosed with WM.
Diagnosis and assessment
Investigations for WM include blood, bone marrow and imaging assessments, as detailed in Table 1[22]. The hallmark of WM is the presence of bone marrow clonal lymphoplasmacytic cells and an IgM paraprotein. Other tests are standard or optional as indicated[22].
Prognosis
The international prognostic scoring system for WM (ISSWM) is based on the assessment of five variables, including age (>65 years), haemoglobin (Hb [<11g/L]), platelets (< 100 x 109/L), beta-2 microglobulin (B2M [>3mg/L]) and IgM paraprotein (>70g/L) — see Table 2[23]. This scoring system predates the use of novel agents, such as ibrutinib and proteasome inhibitors. The ISS should be recorded in clinical trials but otherwise should not influence treatment decisions.
Differential diagnosis
Diseases that have similar characteristics to WM include IgM-multiple myeloma, marginal zone lymphoma (MZL), mantle cell lymphoma and other IgM-secreting lymphomas. Discriminating features include clinical, morphological, cytogenetic and molecular features[19].
Management
Patients with asymptomatic disease should be managed with an active surveillance strategy, which includes monitoring of clinical and laboratory findings every three to six months and appropriate immunisations (as listed below). The median time for patients to progress to symptomatic disease is approximately five years and the main risk factors for progression are IgM paraprotein concentration, degree of bone marrow infiltration with lymphoplasmacytic cells and haemoglobin value[24].
Indications for treatment include clinical and laboratory parameters. Clinical indications include constitutional symptoms (fever, night sweats, weight loss, severe fatigue), symptomatic hyperviscosity, symptomatic lymphadenopathy or hepatosplenomegaly, or peripheral neuropathy related to the IgM paraprotein. Laboratory indications include cytopenias related to bone marrow infiltration (Hb< 100g/L, Plts<100 x 109/L), symptomatic cryoglobulinaemia, symptomatic cold agglutinin anaemia, autoimmune haemolytic anaemia/thrombocytopenia, WM-related AL amyloid and WM-related nephropathy[19]. Serum IgM level>60g/L at diagnosis is associated with a median time to symptomatic hyperviscosity of three months and should therefore be considered as an indication to start treatment[25]. Patients with WM frequently present with iron deficiency anaemia, which should be investigated and treated according to national guidance[26]. However, iron deficiency in WM may be related to elevated hepcidin levels and those with severely depressed transferrin saturation (<10%) may respond to parenteral iron[27].
Once patients have symptoms of disease, consideration should be given to patient-related factors, including fitness to receive treatment, comorbidities and preference for fixed-duration versus continuous treatment, and to disease-related characteristics, including the tempo of the disease and its molecular features. In all instances, patients should be considered for appropriate clinical trials[19].
Patients with hyperviscosity syndrome should be managed with urgent plasmapheresis followed by systemic therapy[19,28].
Owing to the rarity of WM, therapeutic options are usually based on single-arm phase 2 studies. Clinicians should consult up-to-date clinical guidelines, including the latest guideline from the British Society of Haematology[29].
In the absence of appropriate clinical trials, upfront treatment options include rituximab in combination with chemotherapy or bortezomib, or a BTK-I. Treatment options at relapse include BTK-Is, rituximab- or bortezomib-containing regimens[29]. Second-generation and novel non-covalent BTK inhibitors show a favourable toxicity profile, and the latter may overcome BTK-resistance[30–32]. Inhibition of BCL2, an essential regulator of apoptosis in normal and malignant cells, which is overexpressed in WM lymphoplasmacytic cells, has shown promising in-vitro activity and is being tested in clinical trials[33–35]. Alternative treatment options include proteasome inhibition. Bispecific monoclonal antibodies and CAR-T cell therapies are undergoing investigation[36,37].
Autologous stem cell transplantation remains a second-line option for selected patients, achieving at least a partial response[29].
Patients considered to be unfit to receive chemo-immunotherapy may be treated with oral chlorambucil or rituximab monotherapy (four to eight doses per month). However, caution must be applied with rituximab (monotherapy or in combination with chemotherapy) owing to the risk of IgM flare, which in 40–50% of cases will induce rapid increases in serum IgM, ranging from 25% to 300%, and could worsen hyperviscosity symptoms[38]. The risk of IgM flare may be mitigated by offering pre-emptive plasmapheresis. The reduced toxicity of monotherapy is also offset by a median time to response of 4 months and a shorter progression-free survival (PFS) of 23.1 months[39].
Owing to the nature of the condition, WM patients are likely to receive several therapeutic options during their treatment journeys. Listen to Harriet’s experience to hear the patient’s perspective.
Response assessment
Response to treatment should be assessed using standard criteria (see Table 3)[40]. Note that best responses may be delayed by several months following completion of treatment.
Frontline treatment options
Dexamethasone in combination with rituximab and cyclophosphamide (DRC) (six 28-day cycles) has an objective response rate of 83% and median PFS of 35 months[41]. Median time to response was 4 months. The combination is safe and generally very well tolerated, with the main toxicities being cytopenias (9% grade 3 neutropenia)[42]. Owing to the risk of hyper-IgM syndrome, rituximab is deferred or plasma exchange utilised upfront to ensure that the IgM paraprotein measures <40g/L before rituximab dosing[43].
Bendamustine with rituximab (BR) is favoured in those needing rapid disease control (e.g. in patients needing rapid reduction in IgM paraprotein, management of cryoglobulinaemia or for management of bulky disease). In a frontline study that randomised patients with indolent non-hodgkin’s lymphoma between BR and RCHOP (rituximab, cyclophosphamide, vincristine and prednisolone), BR was superior in patients with WM, with a median PFS of 69.5 months[44]. However, BR comes with an increased toxicity profile of grade ¾ neutropenia and grade ¾ infection. This led to frequent dose reductions in the trial. In a retrospective comparison of BR with DRC in both upfront and relapsed setting, BR showed a trend towards superior PFS (2-year PFS 88% vs. 61% (P=0.07) in the upfront BR and DRC groups, while 2-year PFS was 66% vs. 53%, (P=0.08) in the relapsed refractory setting[45]. Patients should also be made aware of a small risk of alkylator-associated myeloid malignancy[46].
Rituximab maintenance therapy (monthly administration until toxicity or progressive disease) has been studied in patients following BR induction and has not been demonstrated to afford any progression-free or survival advantage[47]. However, consolidative rituximab leads to deepening of response in one third of WM patients after completing rituximab-containing regimens and may be offered to those who have not achieved a very good partial response (VGPR) and to those whose chemoimmunotherapy was truncated[48].
Bortezomib (weekly) in combination with rituximab and dexamethasone has been shown to be effective in both upfront and relapsed/refractory settings. In the upfront setting, 85% of patients responded (3% complete response (CR), 65% PR, 17% minor response). After a minimum follow-up of 32 months, median progression-free survival was 42 months. Peripheral neuropathy occurred in 46% of patients (grade 3 in 7% of patients); only 8% of patients discontinued bortezomib owing to neuropathy[49]. Bortezomib should therefore be avoided in patients with WM-associated neuropathy.
Carfilzomib and ixazomib are second-generation proteasome inhibitors that have demonstrated good activity when combined with rituximab and dexamethasone in WM (phase 2 studies)[50,51]. However, they are currently not licensed in the UK for this indication.
Relapsed disease
Bruton’s tyrosine kinase inhibitors
BTK-Is have shown excellent responses in treatment-naïve and relapsed disease and are currently available as a treatment option at first relapse of WM in the UK. In a a Phase II clinical trial, Ibrutinib was the first-in-class BTK antagonist showing excellent efficacy at a continuous dose of 420mg once daily. In the landmark phase 2 study in patients with previously treated WM, the objective response rate (ORR) was 90.5%, and the major response rate was 73.0%[52]. Responses were greatest in those with MYD88L265P, CXCR4WT disease (100% ORR and 91.2% major response rate). Patients with MYD88L265P + CXCR4WHIM mutations had a slightly reduced response rate of 85.7%, and patients with MYD88WT had the lowest response rates at 71.4 %. The responses were rapid (median time to response = four weeks) and durable, with estimated two-year progression-free and overall survival rates among all patients of 69.1% and 95.2%, respectively. The main toxicities were haematological (grade >3 neutropenia 14%; grade >3 thrombocytopenia 13%). However, in a retrospective review of 112 ibrutinib-treated patients with WM (treatment durations = 43 months), 11% experienced atrial fibrillation[53].
Patients with atrial fibrillation should be assessed for stroke risk and considered for anticoagulation with low-molecular-weight heparin or factor Xa inhibitors; vitamin K inhibitors should be avoided given the evidence of excessive bleeding in clinical trials[54].
Toxicities are common and often associated with the off-target effects of the molecule, including increased risk of bleeding and bruising, diarrhoea, arthralgia/myalgia and rash. There is further information on toxicities and their management in the PJ podcast ‘BTK inhibitors: what pharmacists need to know’.
Ibrutinib is metabolised through the cytochrome P450 (CYP450) pathway, therefore, strong inhibitors/inducers of CYP450 should be avoided due to potential drug interactions.
A retrospective study found that ibrutinib-withdrawal symptoms were reported in 20% of patients with WM who were undergoing ibrutinib pause, with fever, body aches, night sweats, and arthralgias being the most common symptoms[55]. Two-thirds of patients had symptoms in the absence of progressive disease, which usually resolved once the drug was resumed[55]. Patients experiencing withdrawal may benefit from a short course of prednisone (10mg twice daily) during the interruption period.
The addition of the anti-CD20 monoclonal antibody rituximab to ibrutinib (versus rituximab-placebo) in a randomised phase 3 study of treatment-naïve and previously treated patients revealed an overall and major response rate of 92% versus 47% (P<0.001) and 72% versus 32% (P<0.001), respectively[56]. This translated into a 30-month PFS advantage of 82% versus 28%. Response rates with ibrutinib–rituximab were similar across different CXCR4 genotypes. Patients with mutated CXCR4 showed faster attainment of major responses, suggesting that this population may benefit from the addition of rituximab to ibrutinib[15].
The second generation BTK-Is, including zanubrutinib and acalabrutinib, have shown excellent activity in the relapsed WM setting and, in November 2021, zanubrutinib became the first of these agents to be approved by the European Medicines Agency for the treatment of WM[57]. Following promising early-phase safety and efficacy data, a multi-centre phase 3 study, which randomised patients between ibrutinib and zanubrutinib, revealed zanubrutinib afforded similar responses to ibrutinib with less off-target toxicity[32]. Although VGPR and CR rates improved with zanubrutinib (28% and 19%, respectively), this did not reach statistical significance (P=0.09). Across the molecular subgroups, median time to major response for both arms was 2.8 months; however, the median times to major response in patients with CXCR4WHIM mutations were 3.1 months in the zanubrutinib arm versus 6.6 months in the ibrutinib arm. Notably, atrial fibrillation was recorded in 15% of patients taking ibrutinib and in 2% of patients taking zanubrutinib, while hypertension was reported in 16% of patients taking ibrutinib and 11% patients of patients taking zanubrutinib. Grade 3 neutropenia was more common with zanubrutinib (20% versus 8%), but this did not translate to an increased number of grade >3 infections[32]. Zanubrutinib may therefore be a preferred option in patients with a significant cardiac history.
Acalabrutinib is a potent targeted BTK antagonist that, in contrast to ibrutinib, has little effect on estimated glomerular filtration rate, Tec, and Src family kinases, or interleukin 2–inducible T-cell kinase signalling[13]. Acalabrutinib has been investigated in a multicentre, single-arm study of treatment-naïve and relapsed-refractory patients[30]. With a median follow-up of 27.4 months, 13 of 14 treatment naïve patients and 86 of 92 relapsed or refractory patients achieved an overall response. Grade 3–4 atrial fibrillation occurred in one (1%) patient and grade 3–4 bleeding occurred in three (3%) patients. The most common serious adverse events were lower respiratory tract infection (n=7;7%), pneumonia (n=7;7%)[30]. Acalabrutinib does not currently have marketing approval for treatment of WM in Europe or the UK.
Pirtobrutinib is a non-covalent BTK-I, which inhibits both wildtype and C481-mutant BTK. It leads to continuous BTK inhibition throughout the dosing interval and has been demonstrated to overcome BTK-resistance[58]. The BRUIN phase 1/2 study across patients with B-cell malignancies reported striking activity in a cohort of heavily pre-treated patients with WM. Of the 78 WM patients, (median prior therapies = 3, of which 61 (78%) had received ≥1 prior BTKi (≥2 BTKi in 13/61 patients, 21%). The major response rate for the 72 response-evaluable patients was 68% (95% CI, 56-79), including 17 VGPRs (24%) and 32 PRs (44%). In the safety cohort of all pirtobrutinib-treated patients with B-cell malignancies (n=725), the most frequent grade ≥3 treatment emergent adverse event was neutropenia (20%, n=143). Low rates of grade ≥3 adverse events of hypertension (3%, n=20), haemorrhage (2%, n=16), and atrial fibrillation/flutter (1%, n=7) were observed. Overall, 15 (2%) patients discontinued treatment owing to a treatment-related adverse events [59]. Pirtobrutinib is not currently licensed for the treatment of WM in Europe or the UK.
BTK-Is should be continued until there is disease progression or unacceptable toxicity. Mechanisms of resistance are best characterised in patients receiving ibrutinib for treatment of chronic lymphocytic leukaemia and involve cysteine-to-serine mutation in BTK and mutations in PLCγ2, which lies downstream of BTK[60]. Patients should be monitored for IgM flare and plans should be put in place for subsequent treatment following BTK discontinuation. Covalent BTK-antagonists should be avoided following the development of resistance to ibrutinib or zanubrutinib, owing to the risk of mutations on the BTK and PLGC2 that predict for cross resistance. However, non-covalent BTK antagonists may be effective[60].
Alternative treatments being investigated
The BCL-2 antagonist venetoclax effectively induces apoptosis of LPL cells in vitro and has been shown to be effective in the treatment of patients with relapsed/refractory WM. In a phase 2 study of 32 patients (all MYD88 L2625P, 16 post BTK-inhibitor), the overall, major, and VGP response rates were 84%, 81%, and 19%, respectively. The major response rate was lower in those with refractory versus relapsed disease (50% vs. 95%; P=0.007). The median progression-free survival was 30 months. CXCR4 mutations did not affect treatment response or progression-free survival[34]. Alternative BCL-2 antagonists are being investigated in mature B-cell lymphomas, including WM[61].
Ibrutinib in combination with venetoclax has been shown to induce rapid and profound responses in patients with previously untreated WM. In a single-arm phase 2 study of 45 patients (100% MYD88 L265P, 32% CXCR4 WHIM ), major response rate was 93%, with VGPR 18 (40%), PR 24 (53%), and minor response 3 (7%)[35]. The median time to major response was 1.9 months and was longer in CXCR4-mutated patients (2.8 v 1.8; P=0.048). However, the study had to be abandoned after reporting a higher than expected rate of ventricular arrhythmia (9%)[35].
The CXCR4 antagonists (ulocuplumab and mavorixafor), in combination with ibrutinib, are being investigated in patients with MYD88L265P and CXCR4WHIM mutations, with promising early-phase results[62,63].
Bispecific antibodies that target the B cell maturation antigen (BCMA) have been shown to be highly effective in patients with relapsed-refractory multiple myeloma and will be investigated in WM, where it has been shown that LPL cells express weak-to-moderate levels of BCMA[36].
CAR-T cell therapy (MB-106) has been granted orphan drug status by the US Food and Drug Administration and is showing promising results in early phase trials. In a single-centre study for patients with RR indolent B-cell lymphoma, six patients with WM received the third-generation, fully human CD20 targeting CAR-T product (MB-106) and four patients underwent response assessment[18]. This was a heavily pre-treated cohort (median 6.5 prior lines, all 4 patients were BTK-I refractory). Toxicity was manageable; no patients experienced grade 3 cytokine release syndrome and there was no immune effector cell-associated neurotoxicity. All patients responded to treatment: 2 achieved CR, 1 PR and 1 MR. Durability of response is awaited[37].
Transplantation
Autologous stem cell transplantation is an option in selected chemo-sensitive patients at second line. In patients for whom this may be an option, drugs that impair stem cell mobilisation (fludarabine and chlorambucil) should be avoided. Allogeneic transplantation is an appropriate option in highly selected patients who are refractory to chemo-immunotherapy and BTK-Is[29].
Supportive care
Patients with WM are at increased risk of infection and should receive appropriate vaccinations and anti-microbial prophylaxis according to the treatment regimen[29].
Prophylaxis against Pneumocystis jirovecii is recommended in patients requiring intensive and/or immunosuppressive treatment, including BTKis. Prophylaxis against herpes simplex virus and herpes zoster virus is recommended in patients requiring intensive, immunosuppressive or bortezomib-based therapy. All patients embarking on a new line of treatment should be tested for past viral infection (HBV, HCV, HIV) and should receive appropriate prophylaxis and monitoring according to specialist hepatologist advice[29].
All patients with WM should be offered seasonal influenza and SARS-CoV-2 vaccinations. They should also be offered pneumococcal vaccination in the form of pneumococcal conjugate vaccine (PCV13) followed by pneumococcal polysaccharide vaccine (PPV23), at least two months later[64]. Patients with WM over the age of 50 years are candidates for the non-live shingles vaccine.[22,29]
Patients with secondary hypogammaglobulinaemia should receive anti-microbial prophylaxis and may be considered for immunoglobulin replacement in the context of recurrent infection despite prophylaxis. Patients receiving purine analogues and bendamustine should receive irradiated blood products for the rest of their lives[29].
Support for patients
Living with a condition such as WM can be distressing for many patients. Listen to Harriet’s advice on some of the concerns patients may have following diagnosis.
Patients may benefit from support at all phases of their disease pathway and should have access to a clinical nurse specialist. Patients can be directed to www.WMUK.org.uk for further information, access to regional and non-regional support groups, access to a support line and information about patient-doctor summits.
Best practice
- Investigate anaemia for all causes, including functional iron deficiency, before considering anaemia to be an indication for WM-targeted treatment;
- Consideration of pre-emptive plasma exchange or omission of rituximab until IgM paraprotein <40g/L;
- Avoid bortezomib in patients with peripheral neuropathy and carfilzomib in patients with cardiac risk factors;
- Be alert for ibrutinib-associated atrial fibrillation and manage with a cardiologist;
- Counsel patients to omit ibrutinib for three to seven days pre-, and two to three days after, a surgical procedure;
- Be alert to the risk of IgM flare following interruption of BTK-I dosing. Symptoms usually resolve on restarting treatment and patients may benefit from a short course of prednisolone;
- Ensure that all patients in treatment receive appropriate antimicrobial prophylaxis.
- 1Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: Consensus Panel Recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Seminars in Oncology. 2003;30:110–5. https://doi.org/10.1053/sonc.2003.50082
- 2Swerdlow S, Campo E, Harris N, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC Press 2008.
- 3What is Waldenstrom’s macroglobulinaemia? WM UK. 2022. https://www.wmuk.org.uk/what-is-waldenstroms-macroglobulinaemia (accessed May 2024)
- 4Groves F, Travis L, Devesa S, et al. Waldenström’s macroglobulinemia: incidence patterns in the United States, 1988-1994. Cancer. 1998;82:1078–81.
- 5Kristinsson SY, Björkholm M, Goldin LR, et al. Risk of lymphoproliferative disorders among first-degree relatives of lymphoplasmacytic lymphoma/Waldenström macroglobulinemia patients: a population-based study in Sweden. Blood. 2008;112:3052–6. https://doi.org/10.1182/blood-2008-06-162768
- 6Kyle RA, Larson DR, Therneau TM, et al. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N Engl J Med. 2018;378:241–9. https://doi.org/10.1056/nejmoa1709974
- 7Waldenstrom Macroglobulinemia. American Cancer Society. 2024. https://www.cancer.org/cancer/types/waldenstrom-macroglobulinemia.html (accessed May 2024)
- 8Kyle RA, Treon SP, Alexanian R, et al. Prognostic markers and criteria to initiate therapy in Waldenstrom’s macroglobulinemia: Consensus Panel Recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Seminars in Oncology. 2003;30:116–20. https://doi.org/10.1053/sonc.2003.50038
- 9Ocio EM, Schop RFJ, Gonzalez B, et al. 6q deletion in Waldenström macroglobulinemia is associated with features of adverse prognosis. Br J Haematol. 2006;136:80–6. https://doi.org/10.1111/j.1365-2141.2006.06389.x
- 10Nguyen-Khac F, Lambert J, Chapiro E, et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenstrom’s macroglobulinemia. Haematologica. 2012;98:649–54. https://doi.org/10.3324/haematol.2012.070458
- 11Treon SP, Xu L, Yang G, et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N Engl J Med. 2012;367:826–33. https://doi.org/10.1056/nejmoa1200710
- 12Hunter ZR, Yang G, Xu L, et al. Genomics, Signaling, and Treatment of Waldenström Macroglobulinemia. JCO. 2017;35:994–1001. https://doi.org/10.1200/jco.2016.71.0814
- 13Kofides A, Demos M, Tsakmaklis N, et al. Alternative Mutations and Isoform Dysregulation in MYD88 in Waldenstrom’s Macroglobulinemia. Blood. 2018;132:1566–1566. https://doi.org/10.1182/blood-2018-99-119419
- 14Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–46. https://doi.org/10.1182/blood-2013-09-525808
- 15Varettoni M, Zibellini S, Defrancesco I, et al. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017;102:2077–85. https://doi.org/10.3324/haematol.2017.172718
- 16Roccaro AM, Sacco A, Jimenez C, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123:4120–31. https://doi.org/10.1182/blood-2014-03-564583
- 17Cao Y, Hunter ZR, Liu X, et al. The WHIM-like CXCR4S338X somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia. 2014;29:169–76. https://doi.org/10.1038/leu.2014.187
- 18Castillo JJ, Gustine J, Meid K, et al. Histological transformation to diffuse large B‐cell lymphoma in patients with Waldenström macroglobulinemia. American J Hematol. 2016;91:1032–5. https://doi.org/10.1002/ajh.24477
- 19Ghobrial IM. Are you sure this is Waldenström macroglobulinemia? Hematology. 2012;2012:586–94. https://doi.org/10.1182/asheducation.v2012.1.586.3798562
- 20Levine T. Peripheral neuropathies in Waldenstrom’s macroglobulinaemia. Journal of Neurology, Neurosurgery & Psychiatry. 2006;77:224–8. https://doi.org/10.1136/jnnp.2005.071175
- 21Castillo JJ, Treon SP. How we manage Bing–Neel syndrome. Br J Haematol. 2019;187:277–85. https://doi.org/10.1111/bjh.16167
- 22Kastritis E, Leblond V, Dimopoulos MA, et al. Waldenström’s macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2018;29:iv41–50. https://doi.org/10.1093/annonc/mdy146
- 23Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenström macroglobulinemia. Blood. 2009;113:4163–70. https://doi.org/10.1182/blood-2008-08-174961
- 24Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenström macroglobulinemia: long-term results. Blood. 2012;119:4462–6. https://doi.org/10.1182/blood-2011-10-384768
- 25Gustine JN, Meid K, Dubeau T, et al. Serum IgM level as predictor of symptomatic hyperviscosity in patients with Waldenström macroglobulinaemia. Br J Haematol. 2017;177:717–25. https://doi.org/10.1111/bjh.14743
- 26Anaemia – iron deficiency. National Institute for Health and Care Excellence. 2023. https://cks.nice.org.uk/topics/anaemia-iron-deficiency/ (accessed May 2024)
- 27Treon SP. How I treat Waldenström macroglobulinemia. Blood. 2015;126:721–32. https://doi.org/10.1182/blood-2015-01-553974
- 28Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and Management of Waldenström Macroglobulinemia. JAMA Oncol. 2017;3:1257. https://doi.org/10.1001/jamaoncol.2016.5763
- 29Pratt G, El‐Sharkawi D, Kothari J, et al. Diagnosis and management of Waldenström macroglobulinaemia—A British Society for Haematology guideline. Br J Haematol. 2022;197:171–87. https://doi.org/10.1111/bjh.18036
- 30Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: a single-arm, multicentre, phase 2 study. The Lancet Haematology. 2020;7:e112–21. https://doi.org/10.1016/s2352-3026(19)30210-8
- 31Mato AR, Shah NN, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. The Lancet. 2021;397:892–901. https://doi.org/10.1016/s0140-6736(21)00224-5
- 32Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136:2038–50. https://doi.org/10.1182/blood.2020006844
- 33Chng WJ, Schop RF, Price-Troska T, et al. Gene-expression profiling of Waldenström macroglobulinemia reveals a phenotype more similar to chronic lymphocytic leukemia than multiple myeloma. Blood. 2006;108:2755–63. https://doi.org/10.1182/blood-2006-02-005488
- 34Castillo JJ, Allan JN, Siddiqi T, et al. Venetoclax in Previously Treated Waldenström Macroglobulinemia. JCO. 2022;40:63–71. https://doi.org/10.1200/jco.21.01194
- 35Castillo JJ, Sarosiek S, Branagan AR, et al. Ibrutinib and Venetoclax in Previously Untreated Waldenström Macroglobulinemia. Blood. 2022;140:564–5. https://doi.org/10.1182/blood-2022-155610
- 36Martens AWJ, Rietveld JM, de Boer R, et al. Redirecting T-cell Activity with Anti-BCMA/Anti-CD3 Bispecific Antibodies in Chronic Lymphocytic Leukemia and Other B-cell Lymphomas. Cancer Research Communications. 2022;2:330–41. https://doi.org/10.1158/2767-9764.crc-22-0083
- 37Shadman M, Yeung C, Redman MW, et al. P1097: CD20 CAR-T THERAPY WITH MB-106 FOR BTK INHIBITOR-REFRACTORY WALDENSTRÖM MACROGLOBULINEMIA (WM)/ LYMPHOPLASMACYTIC LYMPHOMA (LPL) – SINGLE INSTITUTION STUDY. HemaSphere. 2023;7:e68877ca. https://doi.org/10.1097/01.hs9.0000971284.68877.ca
- 38Treon SP, Branagan AR, Hunter Z, et al. Paradoxical increases in serum IgM and viscosity levels following rituximab in Waldenstrom’s macroglobulinemia. Annals of Oncology. 2004;15:1481–3. https://doi.org/10.1093/annonc/mdh403
- 39Gertz MA, Abonour R, Heffner LT, et al. Clinical value of minor responses after 4 doses of rituximab in Waldenström macroglobulinaemia: a follow‐up of the Eastern Cooperative Oncology Group E3A98 trial. Br J Haematol. 2009;147:677–80. https://doi.org/10.1111/j.1365-2141.2009.07892.x
- 40Owen RG, Kyle RA, Stone MJ, et al. Response assessment in <scp>W</scp>aldenström macroglobulinaemia: update from the <scp>VI</scp>th <scp>I</scp>nternational <scp>W</scp>orkshop. Br J Haematol. 2012;160:171–6. https://doi.org/10.1111/bjh.12102
- 41Kastritis E, Gavriatopoulou M, Kyrtsonis M-C, et al. Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenström macroglobulinemia: final analysis of a phase 2 study. Blood. 2015;126:1392–4. https://doi.org/10.1182/blood-2015-05-647420
- 42Dimopoulos MA, Anagnostopoulos A, Kyrtsonis M-C, et al. Primary Treatment of Waldenström Macroglobulinemia With Dexamethasone, Rituximab, and Cyclophosphamide. JCO. 2007;25:3344–9. https://doi.org/10.1200/jco.2007.10.9926
- 43Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s Macroglobulinemia. Blood. 2016;128:1321–8. https://doi.org/10.1182/blood-2016-04-711234
- 44Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. The Lancet. 2013;381:1203–10. https://doi.org/10.1016/s0140-6736(12)61763-2
- 45Paludo J, Abeykoon JP, Shreders A, et al. Bendamustine and rituximab (BR) versus dexamethasone, rituximab, and cyclophosphamide (DRC) in patients with Waldenström macroglobulinemia. Ann Hematol. 2018;97:1417–25. https://doi.org/10.1007/s00277-018-3311-z
- 46Martin P, Chen Z, Cheson BD, et al. Long‐term outcomes, secondary malignancies and stem cell collection following bendamustine in patients with previously treated non‐Hodgkin lymphoma. Br J Haematol. 2017;178:250–6. https://doi.org/10.1111/bjh.14667
- 47Rummel MJ, Lerchenmüller C, Hensel M, et al. Two Years Rituximab Maintenance Vs. Observation after First Line Treatment with Bendamustine Plus Rituximab (B-R) in Patients with Waldenström’s Macroglobulinemia (MW): Results of a Prospective, Randomized, Multicenter Phase 3 Study (the StiL NHL7-2008 MAINTAIN trial). Blood. 2019;134:343–343. https://doi.org/10.1182/blood-2019-121909
- 48Castillo JJ, Gustine JN, Keezer A, et al. Deepening of response after completing rituximab‐containing therapy in patients with Waldenstrom macroglobulinemia. American J Hematol. 2020;95:372–8. https://doi.org/10.1002/ajh.25712
- 49Dimopoulos MA, García-Sanz R, Gavriatopoulou M, et al. Primary therapy of Waldenström macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN). Blood. 2013;122:3276–82. https://doi.org/10.1182/blood-2013-05-503862
- 50Treon SP, Tripsas CK, Meid K, et al. Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström’s macroglobulinemia. Blood. 2014;124:503–10. https://doi.org/10.1182/blood-2014-03-566273
- 51Castillo JJ, Meid K, Flynn CA, et al. Ixazomib, dexamethasone, and rituximab in treatment-naive patients with Waldenström macroglobulinemia: long-term follow-up. Blood Advances. 2020;4:3952–9. https://doi.org/10.1182/bloodadvances.2020001963
- 52Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N Engl J Med. 2015;372:1430–40. https://doi.org/10.1056/nejmoa1501548
- 53Gustine JN, Meid K, Dubeau TE, et al. Atrial fibrillation associated with ibrutinib in Waldenström macroglobulinemia. American J Hematol. 2016;91. https://doi.org/10.1002/ajh.24366
- 54Shatzel JJ, Olson SR, Tao DL, et al. Ibrutinib‐associated bleeding: pathogenesis, management and risk reduction strategies. Journal of Thrombosis and Haemostasis. 2017;15:835–47. https://doi.org/10.1111/jth.13651
- 55Castillo JJ, Gustine JN, Meid K, et al. Ibrutinib withdrawal symptoms in patients with Waldenström macroglobulinemia. Haematologica. 2018;103:e307–10. https://doi.org/10.3324/haematol.2017.186908
- 56Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 Trial of Ibrutinib plus Rituximab in Waldenström’s Macroglobulinemia. N Engl J Med. 2018;378:2399–410. https://doi.org/10.1056/nejmoa1802917
- 57Brukinsa. European Medicines Agency. 2023. https://www.ema.europa.eu/en/medicines/human/EPAR/brukinsa#ema-inpage-item-authorisation-details (accessed May 2024)
- 58Mato A, Shah N, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397:892–901.
- 59Palomba ML, Patel MR, Eyre TA, et al. Efficacy of Pirtobrutinib, a Highly Selective, Non-Covalent (Reversible) BTK Inhibitor in Relapsed / Refractory Waldenström Macroglobulinemia: Results from the Phase 1/2 BRUIN Study. Blood. 2022;140:557–60. https://doi.org/10.1182/blood-2022-159123
- 60Woyach JA, Furman RR, Liu T-M, et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N Engl J Med. 2014;370:2286–94. https://doi.org/10.1056/nejmoa1400029
- 61Li C, Wei J, Zhou K, et al. A phase 1 study evaluating the safety, tolerability, pharmacokinetics, and preliminary antitumor activity of Bcl-2 inhibitor BGB-11417 in adult patients with mature B-cell malignancies. JCO. 2023;41:7558–7558. https://doi.org/10.1200/jco.2023.41.16_suppl.7558
- 62Treon SP, Meid K, Hunter ZR, et al. Phase 1 study of ibrutinib and the CXCR4 antagonist ulocuplumab in CXCR4-mutated Waldenström macroglobulinemia. Blood. 2021;138:1535–9. https://doi.org/10.1182/blood.2021012953
- 63Treon SP, Buske C, Thomas SK, et al. Preliminary Clinical Response Data from a Phase 1b Study of Mavorixafor in Combination with Ibrutinib in Patients with Waldenström’s Macroglobulinemia with MYD88 and CXCR4 Mutations. Blood. 2021;138:1362–1362. https://doi.org/10.1182/blood-2021-144706
- 64Pneumococcal: the green book, chapter 25. UK Health Security Agency. 2023. https://www.gov.uk/government/publications/pneumococcal-the-green-book-chapter-25 (accessed May 2024)
1 comment
You must be logged in to post a comment.


Excellent article - extremely comprehensive and detailed with regard to responses to treatment.