CDC / Janice Haney Carr
In short
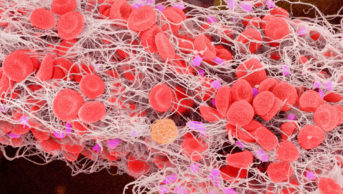
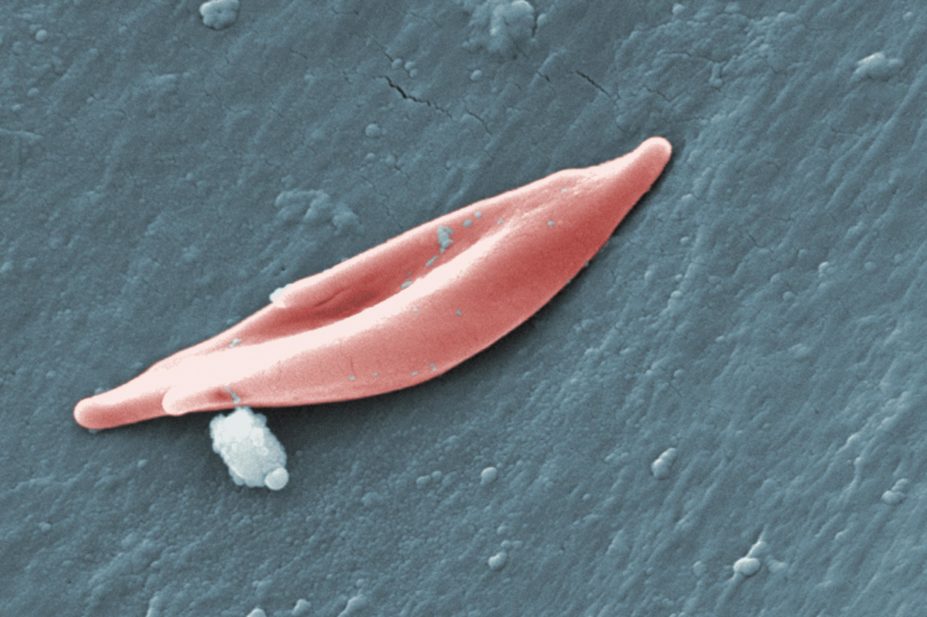
Sickle cell disease is an autosomal recessive genetic condition resulting from the presence of a mutated form of haemoglobin (haemoglobin S), which polymerises under low oxygen conditions, causing red blood cells to become sickle-shaped. These cells cause other cells to become activated in processes similar to vasculopathies. Subsequently, the tissue loses blood supply and the resultant ischaemia causes infarction.
Clinical manifestations do not usually occur until 6–12 months on account of protection against haemoglobin S by the persistence of foetal haemoglobin in the newborn. The most common manifestations include painful vaso-occlusive crises, haemolytic anaemia and end organ damage caused by vasculopathy and tissue ischaemia. Complications may be of sudden onset (sickle cell crisis), but a degree of sickling occurs most of the time, and it is this that leads to long-term organ damage.
Although sickle cell disease is a life-shortening condition, the use of vaccination and antibiotic prophylaxis schedules, transfusion and chelation therapy, as well as the targeted prescription of hydroxycarbamide, have contributed to patients living longer, often with attendant comorbidities.
Sickle cell disease is an autosomal recessive genetic condition resulting from the presence of a mutated form of haemoglobin. It is the most common inherited blood disorder in the UK, affecting 12,000–15,000 people with approximately 250,000 carriers of the sickle cell gene[1]
. It is most common in people of West African or Caribbean descent[2]
.
The cause
of sickle cell disease is a single nucleotide substitution on the beta globin gene on the short arm of chromosome 11. The resulting protein, haemoglobin S (HbS), polymerises under low oxygen conditions, causing red blood cells to become rigid and sickle-shaped. These sickled cells cause
a
ctivation of other circulating cells, including neutrophils, and blood vessel walls in processes similar to those observed in vasculopathies. Su
bsequently, the tissue loses its blood supply and the resultant ischaemia causes tissue infarction[3]
. Heter
ozygosity (one HbS allele and one HbA allele — known as ‘sickle trait’) is thought to confer a certain level of resistance to
Plasmodium falciparum
malaria and may explain the persistence of the HbS gene in areas where malaria is, or was, endemic. The protective effect sickle trait confers against malaria does not extend to homozygous patients, and they still require antimalarial prophylaxis when travelling to e
ndemic areas[3]
.
I
n the UK, all babies are screened for the condition shortly after birth as part of the newborn blood spot t
est[4]
. In add
ition, antenatal screening is offered if the parents are considered at risk of being carriers of the HbS gene because of their ethnicity.
Althou
gh sickle cell disease is still a life-shortening condition, the use of rigorous vaccination and antibiotic prophylaxis schedules, transfusion and chelation therapy and the targeted prescription of hydroxycarbamide have all contributed to patients living longer, although often with attendant mo
rbidities[5]
.
Symptoms
The clinical manifest
ations of sickle cell disease do not usually appear until the age of 6–12 months. This is bacause a level of protection is conferred against the HbS induced abnormalities by the persistence of foetal haemoglobin (HbF) in the newborn.
The most common manifestations are painful vaso-occlusive crises, haemolytic anaemia and end organ damage caused by vasculopathy and tissue ischaemia. Complications may be of sudden onset, known as a sickle cell crisis, but a degree of sickling occurs most of the time, and it is this that leads to long-t
erm organ damage.
Vaso-occlusive crises
result from obstruction of the microv
asculature by sickled red blood cells, causing ischaemia and pain. The pain, typically presenting in the chest, back or extremities, can be excruciating and is one of the most distressing consequences of sickle cell disease. The pain can last for a few hours up to several days. When the patient is otherwise well and pain is the only feature, it is known as a simple vaso-occlusive crisis. A vaso-occlusive crisis can happen in a particular organ, for example the lungs causing a chest syndrome, the brain causing a stroke or the penis causing priapism.
Crises may occur for no obvious reaso
n, or there may be a precipitating trigger factor. Common trigger factors include dehydration, a sudden change in body temperature, for example caused by infection or en
vironmental factors, or hypoxia caused by stress or exercise[6]
.
Patients with sickle cell disease
should be advised to avoid excess alcohol, smoking and illicit drug use, all of which are potential triggers.
Anaemia
associated with sickle cell disease is chronic and caused by intravascular haemolysis resulting in a reduced lifespan of the ab
normal red blood cells (10–20 days compared with 100–120 days in a healthy adult
[7]
). It may be complicated by megaloblastic changes caused by folate deficien
cy[8]
. Childre
n with sickle cell disease may compensate for the anaemia with an increasing heart rate and stroke volume, but often suffer from reduced stamina when taking part in physical exercise at school. Anaemia will worsen during a vaso-occlusive crisis, as the haemolytic rate increases. Anaemia can also worsen in the presence of Parvovirus B19 infection and if the blood pools in the liver or spleen, known as sequest
ration[9]
.
Micro-infarcts
caused by sickling most commonly affect the spleen, kidney, skeleton and central nervous system. Obstruction
of the microvasculature in the spleen renders patients functionally asplenic and susceptible to infection, particularly with encapsulated bacteria such as
Streptococcus pneumonia, Escherichia coli, Haemophilus
and
Meningococcus
[10]
.
Repeated in
farction of the bone occurs when sickled red blood cells become lodged in the arterial and venous sinusoids of the skeleton. Over time, this can lead to bone and joint destruction, particularly in weight-bearing regions, and can potentially culminate in avascular necrosis of the femoral or humeral head. It is not uncommon for patients to need a joint replacement at an early
age[11]
.
Severe complications
include stroke, acute chest syndrome and pulmonary hypertension.
Stroke
affects approximately 10% of patients before 20 years of age. The incidence reduces bet
ween the ages of 20 years and 29 years, but is subsequently higher in o
lder patients[12]
. There may be asso
ciated convulsions or other neurological deficit.
Acute chest syndrome
is a medical emergency chara
cterised by fever and/or respiratory symptoms, and an accompanying pulmonary infiltrate on a chest X-ray. The cause of acute chest syndrome may be infective or caused by infarcts either in the lungs or in the bone marrow leading to a fat embolus. The first sign is usually a mild
hypoxia.
Pulmonary hypertension
associated with sickle cell disease may occur as a res
ult of increased levels of free plasma haemoglobin caused by haemolysis leading to reduced availability of nitric oxide, but the precise cau
se is still unknown[13]
.
Renal disease
is increasingly recognised as a cause of long-ter
m morbidity as this patient cohort ages, although end stage renal failure can be seen in younger patie
nts[14]
.
Other long-term complications of sickle cell disease include vascular changes in the retina[15]
, le
g ulcers
[16]
which are painful and
slow to heal, cholelithi
asis[17]
caused by hyperbilirubinaemia f
rom chronic haemolysis, g
rowth retardation and delayed puberty[18]
.
Management
The aim of treatment for patients with sickle cell disease is symptom control and the prevention or management of complications. Allogeneic stem cell transplantation is the only curative therapy available for sickle cell disease and has been used with some success[19]
,[20]
. However, its wider application is limited by the availability of suitable donors and the relatively high treatment related mortality, making it hard to define for which patients the procedure has an acceptable risk-benefit profile.
Hydroxycarbamide
is the only approved disease-modifying drug available for sickle cell disease. It has multiple beneficial effects, including increasing the proportion of HbF in red blood cells, thereby reducing the propensity for them to sickle and decreasing neutrophil counts. It has been shown to reduce the frequency of vaso-occlusive episodes and acute chest syndrome in children and adults[21]
,[22]
. Close monitoring of the patient’s full blood count is required during therapy to promptly identify excessive myelosuppression.
Blood transfusions
may serve two purposes in a patient with sickle cell disease. Simple top-up transfusions help
to correct the underlying anaemia and should be performed if clinically indicated and not just to correct low haemoglobin levels, as repeated top-ups will lead to iron overload and/or alloimm
unisation[23]
.
Exchange transfu
sions involve removing some of the patient’s own blood at the time of transfusion, thereby lowering the percentage of HbS relative to HbA. Exch
ange transfusions may be used to t
reat or prevent the occurrence of vaso-
occlusive complications such as stroke[24]
. Automated exchange transfusions using an
apheresis machine have the advantage of being iron neutral, thereby reducing the need for chelation therapy.
Although the evidence base for it is lacking, patients are also usually prescribed folic acid supplementation, 5mg d
aily[25]
.
Managing vaso-occlusive crisis
Vaso-occlusive crises are the most common reason for hospital admission in patients with sickle cell disease. The National Institute for Health and Care Excellence has produced a clinical guideline[26]
containing management recommendations, which in the first instance usually requires the rapid initiation of opioid analgesia to manage pain. This can be given orally but it may need to be administered parenterally until pain is controlled.
Dia
morphine is more water soluble and higher doses may be diluted in smaller volumes, thus allowing subcutaneous administration in patien
ts who often have poor venous access because of many years of repeated cannulation[6]
. However, if intravenous access
is available then morphine is preferable. Intranasal diamorphine has also been used in
the emergency department setting[27]
.
It is common pra
ctice to continue treatment for the initial 24 hour period with hourly bolus administrations to establish a baseline requirement that may then be converted into an infusion for patient controlled analgesia (PCA).
The opioid requirements may be high, with patients requiring doses sometimes running into hundre
ds of milligrams each day[6]
, particularly during the initial phase of a crisis. Once a safe and effectiv
e analgesic regimen is established, conversion to an oral regimen in preparation for discharge and self-management may be considered.
High dos
e opioid treatment can be associated with side effects including pruritus, which can be managed with sedating antihistamines (e.g. hydroxyzine), and nausea, which can be managed with antiemetics. Combination laxative treatment is often required because both opioid therap
y and vaso-occlusive crisis can cause constipation[6]
. Patients will commonly know what regime
n of supportive medication works well for them in relieving these side eff
ects.
Oxycod
one and fentanyl are useful alternatives for patients who are intolerant of diamorphine and morphine despite the use of relieving measures. A small proportion of crises with more moderate pain may be managed using simple oral analgesics, such as paracetamol and codein
e.
Chelation therapy may be required in patients with sickle cell disease. With the absence of a physiological mechanism for excreting excess iron, repeated top up or manual exchange blood transfusions will lead to a state of iron overload. Labile iron generates free radicals and causes often irreversible tissue damage. Initially, iron derived from the breakdown of the transfused red cells accumulates in macrophages of the liver, spleen and bone marrow. Subsequent accumulation in hepatocytes may lead to fibrosis and liver dysfunction in the short term, potentially leading eventually to cirrhosis, liver failure and hepatocellular carcinoma. Iron deposition in the heart may lead to arrhythmias and ventricular failure but this is extremely rare in sickle cell disease. Endocrine tissues may also be affected, with damage to the pancreas causing diabetes, the thyroid causing hypothyroidism, the parathyroid causing hypoparathyroidism and the anterior pituitary causing poor growth and sexual development[28]
.
The aim of iron chelation thera
py is to maintain iron balance and prevent iron-induced tissue damage. There are three iron-chelating drugs currently available: desferrioxamine, deferasirox and deferiprone.
Desferrioxamine is given parenterally and because of its short half-life and the limited amount of free iron available to be bound at any one time, it needs to be given as a series of prolonged infusions. The dose is variable, depending on the degree of iron overload and the degree of any pre-existing tissue damage.
The usual dose range is 20–60mg/kg/day (20–40mg/kg/day if the patient is still growing) given as a subcutaneous infusion over 8–12 hours on five to seven days per week. Patients may choose to prepare the infusions at home and administer via a syringe driver. Alternatively, many centres have sterile compounding and home delivery arrangements to facilitate patient compliance with treatment.
Injection site reactions
may be managed by adding a small dose of hydrocortisone to the infusion solution. Other side effects of desferrioxamine include retinal toxicity, high frequency hearing loss and an increased susceptibility to systemic
Yersinia
spp. infection[29]
because of its potential to act as a bact
erial sider
ophore.
Deferasirox (Exjade; Novartis) is taken as a once daily tablet, with a usual dose range of 20–40mg/kg/day. The tablets are dispersible, which facilitates administration to young children. It must be taken once daily on an empty stomach at least 30 minutes before food. The tablets are dispersed by stirring in a glass of water or orange or apple juice (100–200ml) until a suspension is obtained. After the suspension has been swallowed, any residue must be re-suspended in a small volume of water or juice and swallowed. Side effects of deferasirox include dose-dependent increases in serum creatinine (36% of patients), gastrointestinal disturbances (26% of patients) and skin rash (7% of patients)[30]
.
Deferiprone is usually reserved for patients where initial therapy with desferrioxamine[31]
or defera
sirox is inadequate or is not tolerated. Careful monitoring of the patient’s full blood count is required because of the small risks of neutropenia and agranulocyt
osis.
Common p
ractice is to initiate iron chelation therapy after 20 transfusion episodes or when serum ferritin exceeds 1000µg/L. Starting treatment at lower levels of iron loading increases the risk of toxicity with desferrioxamine and, although not confirmed, this may be a risk with other chelato
rs.
Prophylactic antibiotics
, usually twice daily phenoxymethylpenicillin, are required for life in p
atients with sickle cell disease because of the risk of infection with encapsulated bacteria. Erythromycin is a suitable alternative in patients with p
enicillin allergy.
Patients s
hould receive the routine childhood vaccination schedule that includes prevenar. It is particularly important that they are vaccinated against Haemophilus influenzae type B (HiB), meningococcal ACWY and B strains, hepatitis B and pneumococci. Patients should be re-immunised with a pneumococcal vaccine every fi
ve years and should receive an annual influenza vaccination[32]
.
Patients will need additional vaccinations for foreign travel, such as hepatitis A (depending on the destination) and travel insurance that covers the costs of repatriation. Patients must take antimalarial prophylaxis when travelling to endemic areas and should know their G6PD status when deciding on appropriate therapy.
Etilefrine
is an alpha-adrenergic agonist used for the prevention and treatment of priapism[33]
,
[34]
.
Pseu
doephedrine
and oral
terbutaline
are alternative options. Surgical intervention may be required for cases that f
ail to respond to drug therapy
[35]
.
Future options
A number of disease modifying agents are undergoing investigation but none are close to routine clinical use. These include direct inhibitors of HbS polymerisation (5-hydroxymethyl furfural[36]
), Gardos channel blockers (clotrimazole[37]
, senicapoc[38]
) and selectin inhibitors that may decrease adhesion between blood cells and the microvasculature (rivipansel[39]
).
Gene therapy has made some promising progress but is not yet a validated treatment. One approach currently being tested involves the transplantation of autologous haematopoietic stem cells transduced
ex vivo
with a lentiviral beta globin gene carrying vector[40]
.
Jade
nu (Novartis)
is a new formulation of deferasirox recently approved by the US Food and Drug Admin
istration[41]
that will si
mplify administration of the drug. It is a film-coated tablet that may be swallowed whole and taken once daily with or without a light meal.
Simon Cheesman is lead ph
armacist at University College London Hospital (UCLH) NHS Foundation Trust.
References
[1] National Health Service (NHS) choices. Sickle cell anaemia. Available at: www.nhs.uk/conditions/Sickle-cell-anaemia/Pages/Introduction.aspx (accessed August 2015).
[2] Modell B & Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization 2008;8(6):480–487.
[3] Oluseyi O & Aika O. Malaria chemoprophylaxis in sickle cell disease. Cochrane Database Syst Rev. 2006.
[4] National Health Service (NHS) Population screening programmes. NHS newborn blood spot (NBS) screening programme. Available at: www.gov.uk/topic/population-screening-programmes/newborn-blood-spot (accessed August 2015).
[5] Platt OS, Brambilla DJ, Rosse WF et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–1644.
[6] Rees D et al. on behalf of the British Committee for Standards in Haematology General Haematology Task Force by the Sickle Cell Wo rking Party. Gui delines for the management of the acute painful crisis in sickle cell disease. British Journal of Haematology 2002;120:744–752.
[7] McCurdy PR. 32DFP and 51Cr for Measurement of Red Cell Life Span in Abnormal Hemoglobin Syndromes. Blood 1969;33(2):214–224.
[8] Liu YK. Folic acid deficiency in sickle cell anaemia. Scand J Haematol 1975;14(1):71–79.
[9] Mallouh AA & Qudah A. Acute splenic s equestration together with aplastic crisis caused by human parvovirus B19 in patients with sickle cell disease. J Pediatr 1993;122(4):593–595.
[10] Davies JM, Lewis MP, Wimperis J et al. Review of g uidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen: Prepared on behalf of the British Committee for Standards in Haematology by a Working Party of the Haemato-Oncology Task Force. British Journal of Haematology 2011;155:308 –317.
[11] MartÃ-Carvajal AJ, Solà I & Agreda-Pérez LH. Treatment for avascular necrosis of bone in people with sickle cell disease. Cochrane Database Syst Rev 2012;16:5.
[12] Ohene-Frempong K, Weiner SJ, Sleeper LA et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998;91:288–294.
[13] Bunn H, Nathan DG, Dover GJ et al. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood 2010:116(5);687–692.
[14] Saborio P & Scheinman J. Sickle Cell Nephropathy. Journal of the American Society of Nephrology 1999;10(1):187–192.
[15] Downes SM, Hambleton IR, Chuang EL et al. Incidence and natural history of proliferative sickle cell retinopathy. Ophthalmology 2005;112(11):1869–1875.
[16] Minniti CP, Eckman J, Sebastiani P et al. Leg ulcers in sickle cell disease. Am J Hematol. 2010;85(10):831–833.
[17] Stephens CG & Scott RB. Cholelithiasis in sickle cell anemia: surgical or medical management. Arch Intern Med 1980;140(5):648–651.
[18] Rhodes M, Akohoue SA, Shankar SM et al. Growth patterns in children with sickle cell anemia during puberty. Pediatr Blood Cancer 2009;53(4):635–641.
[19] Bernaudin F, Socie G, Kuentz M et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007;110:2749–2756.
[20] Hsieh MM, Kang EM, Fitzhugh CD et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009;361:2309–2317.
[21] Thornburg CD, Files BA, Luo Z et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood 2012;120(22):4304–4310.
[22] Voskaridou E, Christoulas D, Bilalis A et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle-cell syndromes: results of a 17-year, single center trial (LaSHS). Blood 2010;115(12):2354–2363.
[23] Smith-Whitley K & Thompson AA. Indications and complications of transfusions in sickle cell disease. Pediatr Blood Cancer 2012;59(2):358–364.
[24] Kassim AA, Galadanci NA, Pruthi S et al. How I treat and manage strokes in sickle cell disease. Blood 2015;125(22):3401–3410.
[25] Al-Yassin A, Osei A & Rees D. Folic acid supplementation in children with sickle cell disease. Arch Dis Child 2012;97:A91–A92.
[26] National Institute for Health and Care Excellence. Sickle cell acute painful episode: management of an acute painful sickle cell episode in hospital. (2012) . NICE clinical guideline 143.
[27] Regan L, Chapman AR, Celnik A et al . Nose and vein, speed and pain: comparing the use of intranasal diamorphine and intravenous morphine in a Scottish paediatric emergency department. Emerg Med J 2013;30(1):49–52.
[28] Fleming RE & Ponka P. Iron overload in human disease. N Engl J Med 2012;366:348–359.
[29] Robins-Browne RM & Prpic JK. Effects of iron and desferrioxamine on infections with Yersinia enterocolitica. Infect Immun 1985;47(3):774–779.
[30] Exjade. Summary of Product Characteristics. Available at: www.medicines.org.uk/emc/medicine/18805 (accessed August 2015).
[31] Hoffbrand V. Deferiprone therapy for transfusional iron overload. Best Pract Res Clin Haematol 2005;18(2):299–317.
[32] Public Health England. Immunisation against infectious disease. Immunisation of individuals with underlying medical conditions: the green book, chapter 7. May 2014.
[33] Okpala I, Westerdale N, Jegede T et al. Etilefrine for the prevention of priaprism in adult sickle cell disease. Br J Haematol 2002;118(3):918–921.
[34] Gbadoe AD, Atakouma Y, Kusiaku K et al. Management of sickle cell priapism with etilefrine. Arch Dis Child 2001;85(1):52–53.
[35] Cherian J, Rao AR, Thwani A et al . Medical and surgical management of priapism. Postgrad Med J 2006;82(964):89–94.
[36] Evaluation of different dose regimens of Aes-103 given for 28 days to subjects with stable sickle cell disease. NCT01987908. Available at: www.clinicaltrials.gov/ct2/show/NCT01987908 (accessed August 2015).
[37] Brugnara C, Gee B, Armsby CC et al. Therapy with oral clotrimazoleinduces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J Clin Invest 1996;97:1227–1234.
[38] Ataga KI, Smith WR, De Castro LM et al. On behalf of the ICA-17043-05 investigators. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood 2008;111(8):3991–3997.
[39] Telen MJ, Wun T, McCavit TL et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 2015;125(17):2656–2664.
[40] Cavazzana M. Outcomes of gene therapy for B-thalassemia major and severe sickle cell disease via transplantation of autologous hematopoietic stem cells transduced ex vivo with a lentiviral beta globin vector. Abstract S466. European Hematology Association 20th Congress. 11-14 June 2015.
[41] Jadenu® (deferasirox). Prescribing information. Available at: www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf (accessed August 2015).