This content was published in 2004. We do not recommend that you take any clinical decisions based on this information without first ensuring you have checked the latest guidance.
The main challenge in designing a “standard” drug dosage regimen is the variability in drug handling that exists from patient to patient. Understanding the sources of this variability and adjusting drug doses accordingly is an area in which pharmacists can make a major impact on risk management and patient care. There are three main sources of variability: those related to observable clinical characteristics, those that are genetically determined and those related to other drug therapy.
Age
Pharmacokinetic variability is particularly important at the extremes of age. This reflects differences in body composition and function. The weight-related volume of distribution of water-soluble drugs (eg, aminoglycoside antibiotics) is higher in neonates than in adults because of the greater proportion of water per kilogram of body weight. This means that if an adult and a neonate are each given a 5mg/kg dose of a water soluble drug, such as gentamicin, the maximum concentration will be around 16–20mg/L in the adult and 10–11mg/L in the neonate. In contrast, the relative volume of distribution of fat-soluble drugs, such as diazepam, is higher in the elderly. Age also affects drug binding. The binding of drugs to albumin in the plasma is reduced in neonates and in the elderly whereas binding to alpha-1-acid glycoprotein (an “acute phase reactant” that is released in response to inflammation and trauma) is generally low in infants but may be elevated in elderly patients, especially if other disease processes are present.
The clearance of drugs that are eliminated by the kidney is significantly influenced by age. Renal function is low at birth, particularly in premature neonates, and increases dramatically over the first two weeks of life, which means that the dose requirements of renally cleared drugs are highly variable during this period. In later years, renal function progressively declines and may be substantially reduced in elderly patients, despite apparently “normal” serum creatinine concentrations.
Age also affects the hepatic metabolism of drugs. At birth, most drug metabolising enzymes are present but the activity of many enzyme systems is low, so this can result in drug accumulation and toxicity. In contrast, the evidence that elderly patients have significantly reduced hepatic function is limited, and it is often unnecessary to reduce the dose of drugs that are cleared by hepatic metabolism unless other factors (eg, cardiac disease, hepatic disease or drug interactions) are involved.
Body weight
Many drug doses are based on the body weight of the patient (expressed as mg/kg), but the influence of obesity or malnourishment on dosage requirements is not always considered. If a drug is highly water soluble, it will have a limited distribution into fat and its volume may, therefore, be better correlated with “ideal body weight” or lean body mass. In contrast, total body weight may be more relevant for a drug that is highly lipid soluble. However, although renal and hepatic function are related to body size, obesity may not produce a corresponding increase in renal and hepatic function. Consequently, the use of total body weight to determine a drug dosage regimen could result in toxic effects if the patient is grossly obese.
Sex
Although differences in drug handling be tween males and females have been observed for a number of drugs, even after correcting for weight, such differences are often considered to be too small to warrant a dose change. However, doses can differ if the drug is being used for a sex-specific indication, for example, buserelin for endometriosis (300∝g intranasally three times a day) or for prostate cancer (200∝g intranasally six times a day).
Renal function
Because many drugs are partially or mainly cleared by excretion through the kidneys, deterioration in renal function is one of the principal reasons for drug dosage adjustment in clinical practice. As demonstrated with many antibiotics, the increase in elimination half-life may mean that a drug should be given less frequently to a patient with renal impairment. In contrast, the dose, rather than the dosage interval, is usually altered for a drug that has a long elimination half-life, such as digoxin, because it is easier for the patient to take a daily dose than a dose on alternate days.
Another factor that could become important if renal function continues to deteriorate is the influence of renal replacement therapy on drug clearance and dosage requirements. Haemodialysis is an efficient way of removing many drugs, although the relatively short period of dialysis (typically four hours, three times a week) means that the total amount of drug removed is often small. Doses generally correspond to those recommended for a glomerular filtration rate (GFR) less than 10ml/min and the timing of doses is not critical. However, some antibiotics and antifungal drugs are often deliberately given after dialysis to ensure that therapeutic concentrations are maintained. In contrast, continuous procedures, such as haemofiltration, especially the newer forms of haemodiafiltration with high flow rates, produce a higher overall drug clearance and, consequently, dosage recommendations based on a GFR of 10 to 20ml/min are often used.
Liver function
Unlike in renal disease, where creatinine clearance estimates provide a reasonable guide to alterations in drug dosage requirements, indicators of hepatic disease, such as elevated liver enzymes, low serum albumin concentrations and clotting abnormalities, cannot be directly related to drug clearance. Nevertheless, patients with severe cirrhosis will often need reduced doses of hepatically cleared drugs to avoid toxicity. The increased exposure demonstrated in such patients may not simply be due to a decline in hepatic clearance but also, for some drugs, to an increase in bioavailability caused by a reduction in first-pass metabolism. For example, the bioavailabilities of morphine and labetalol have been reported to double in patients with cirrhosis.
Genetic factors
Genetic polymorphisms that lead to the pro duction of isoenzymes with reduced or no activity or to multiple copies of an enzyme with high activity make a major contribution to the variability in the dose requirements of drugs that are eliminated by hepatic metabolism. Of particular note is the cytochrome P4502D6 isoenzyme (CYP2D6), which catalyses the metabolism of many anti arrhythmic, antidepressant and antipsychotic drugs. Genetic variability in transporter proteins, such as P-glycoprotein, the expression of which is controlled by the MDR1 gene, are also increasingly being recognised for their importance to pharmacokinetic variability.
P-glycoprotein is present in many body tissues, where it can influence drug absorption, distribution and elimination. For example, low digoxin bioavailability has been associated with high levels of P-glycoprotein, which reduces bioavailability by pumping the drug back through the intestinal wall into the gut.
In the future, genetic screening may be used to help determine the initial doses of drugs for which genetically determined variability in pharmacokinetics has been characterised.
Drug interactions
Many of the clinically significant interactions between drugs are pharmacokinetic in origin, often due to induction or inhibition of metabolising enzymes or transporter proteins. However, interactions can also occur between drugs and food supplements or herbal remedies.
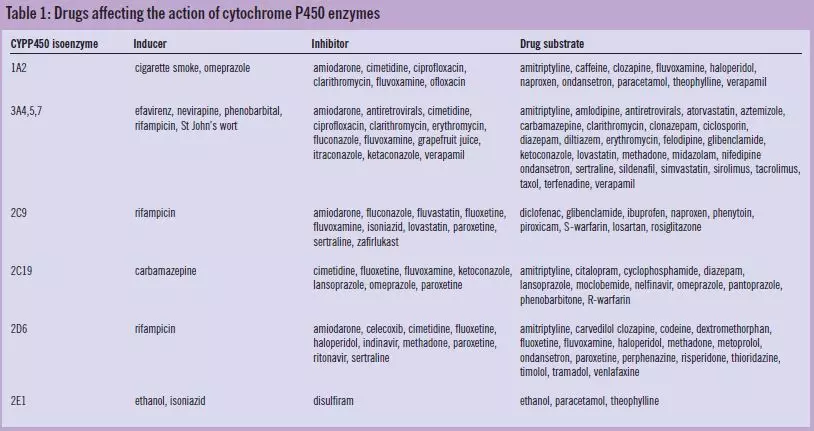
Table 1 contains a list of isoenzymes of CYP450, some of their major inducers and inhibitors and the substrates that are associated with them. Interactions involving competitive inhibition often occur within two to three days whereas induction may take any thing from hours to weeks. If the interacting drug has a long elimination half-life, the interaction may persist for some time after it has been discontinued. It is, therefore, important not only to consider potential interactions when two drugs are given together but also when one is stopped.
Absorption
Recent studies have demonstrated the importance of intestinal CYP3A4 and P-glycoprotein in drug absorption. Induction of these mechanisms by rifampicin and by St John’s wort have been shown to reduce the bioavailability of digoxin. Absorption can also be altered by drug interactions within the gut that result from binding to other drugs, such as cholestyramine or antacids, or to enteral feeds, as in the case of phenytoin.
Distribution
Drug distribution can be altered by interactions that cause displacement from plasma protein binding but these do not normally alter maintenance dose requirements unless there is also a reduction in the clearance of unbound drug.
Clearance
Drug clearance can be altered by enzyme induction or inhibition. Due to the wide variability in enzyme activity, the clinical significance of an interaction is often difficult to predict on an individual basis. Interactions are often dose-dependent and the timescale of the offset and onset of the effect depends on the pharmacokinetics of not only the two (or more) drugs concerned but also the isoenzyme(s) responsible for their metabolism.
Other pharmacokinetic interactions have been reported that involve changes in renal clearance (eg, probenecid reduces the renal clearance of many antibiotics by competing for the anion secretion transport mechanism) and changes in biliary secretion and enterohepatic recirculation (eg, penicillins can impair the recirculation of oral contraceptives by altering the bacterial flora of the gut).
Other factors
A range of other factors, such as pregnancy, cardiac disease, respiratory disease and infection can lead to variability in drug concentration-time profiles. In addition, variations in adherence, which may take the form of missed doses, missed days or simply variations in the times of dosing, will lead to unequal dosage intervals and may affect the patient’s response to therapy. This is a particular problem when the drug has a short elimination half-life and the aim is to maintain concentrations above a minimum value. For example, variability in the dosage intervals of antiretroviral drugs can lead to low concentrations that facilitate the development of drug resistance.
Summary
A range of factors, including clinical characteristics, genetic factors, concomitant disease states and other drug therapy can influence the pharmacokinetics of a drug and consequently the patient’s response to drug therapy. Drug dosage adjustments are often required to ensure that the patient achieves an adequate dose without risk of toxicity.
References
1. Verbeeck RK, Horsmans Y. Effect of hepatic insufficiency on pharmacokinetics and drug dosing. Pharmacy World and Science 1998;20:183–92.
Further reading
- Begg E. Instant Clinical Pharmacology, Blackwell Publishing Ltd., Oxford, 2002
- Bunn R, Ashley C Eds. The Renal Drug Handbook, Second Edition, Radcliffe Medical Press Ltd, Oxon 2004.
- Ritschel WA, Kearns GL. Handbook of Basic
- Pharmacokinetics. 5th Edition. American Pharmaceutical Association, Washington DC, 1999.
- Stockley IH, Ed, Drug Interactions. London: Pharmaceutical Press. Electronic version, 2003.