

Key points
- The advantages and disadvantages of oral rather than intravenous administration of anticancer drugs are summarised.
- The oral bioavailability of many anticancer drugs is low and highly variable because of sub-optimal physicochemical properties or appreciable first-pass metabolism and efflux transport or both.
- Potential solutions to low oral bioavailability include the development of pro-drugs, co-administration with inhibitors of metabolism or efflux transport and formulation in carrier systems.
Cancer learning ‘hub’
Pharmacists are playing an increasingly important role in supporting patients with cancer, working within multidisciplinary teams and improving outcomes.
However, in a rapidly evolving field with numbers of new cancer medicines is increasing and the potential for adverse effects, it is now more important than ever for pharmacists to have a solid understanding of the principles of cancer biology, its diagnosis and approaches to treatment and prevention.
This new collection of cancer content, brought to you in partnership with BeOne Medicines, provides access to educational resources that support professional development for improved patient
Introduction
Changes in the treatment of cancer mean that more drugs can be administered orally as well as through the traditional intravenous route. Most patients would prefer to have oral chemotherapy in their home[1],[2],[3],[4]
. However, while there are distinct advantages in using oral rather than intravenous administration, not the least being greater convenience and less discomfort, there are some significant disadvantages (see Box 1).
Box 1: Advantages and disadvantages of oral anticancer drugs
Advantages
- Convenience (administration in the home);
- Avoidance of discomfort and risks of infection, phlebitis and extravasation of intravenous (IV) lines;
- Avoidance of hypersensitivity to IV vehicles (e.g. Chremophore EL®);
- Avoidance of the costs of hospitalisation;
- Modulation of drug release from the dosage form, affording options for optimising effect–time profile;
- More sustained inhibition of target.
Disadvantages
- Low and highly variable bioavailability, including dose-limited absorption;
- Non-adherence, incomplete adherence or over-adherence;
- Gastrointestinal side effects (e.g. diarrhoea, cytotoxicity, dyspepsia);
- Gastrointestinal disease and surgery;
- Food–drug and drug–drug (e.g. antacid) interactions;
- Limited dose options;
- Potentially decreased contact with the oncology team;
- Increased costs of developing sophisticated formulations; difficulty in establishing bioequivalence (generics).
Incomplete adherence to oral drug therapy in the outpatient setting is a major cause of variability in drug response[5],[6]
, even in people with cancer[1]
. Or, on the other hand, some patients may exceed dosage recommendations in an attempt to become disease-free sooner. Another issue is that many oncologists believe that anticancer drugs are best given intravenously because they require close supervision[1]
. Furthermore, any cost savings associated with avoidance of hospitalisation may well be offset by the increased cost of developing sophisticated oral formulations. It should also be considered how different healthcare systems work, for example in the United States where there can be a lack of reimbursement for home treatment.
While these clinical, behavioural and economic issues are part of the wider discussion of oral versus intravenous chemotherapy, this article will concentrate on pharmacokinetic barriers to the safe and effective use of oral agents. In doing so, it should be noted that there are several extensive prior reviews, which should be consulted for more detailed information[7],[8],[9]
.
The importance of understanding pharmacokinetic properties of oral anticancer therapy
An important class of targeted anticancer drugs is the small molecule tyrosine kinase inhibitors (sTKIs). They are now used widely for cancer chemotherapy and exemplify the need to understand pharmacokinetic properties as a major determinant of successful oral therapy. Although sorafenib showed a three-month survival benefit in phase III clinical trials, a study of survival in real-world patients with advanced hepatocellular carcinoma indicated ineffective treatment (see Figure 1). While this outcome was attributed to more severe disease and worse liver function in the latter patients, additional possibilities include the use of a fixed-dose, tumour heterogeneity, non-adherence and, importantly, highly variable pharmacokinetics (including absorption) — a hallmark of TKIs[9]
. Close monitoring of the exposure to these drugs with dose adjustment is essential to their safe and effective use.
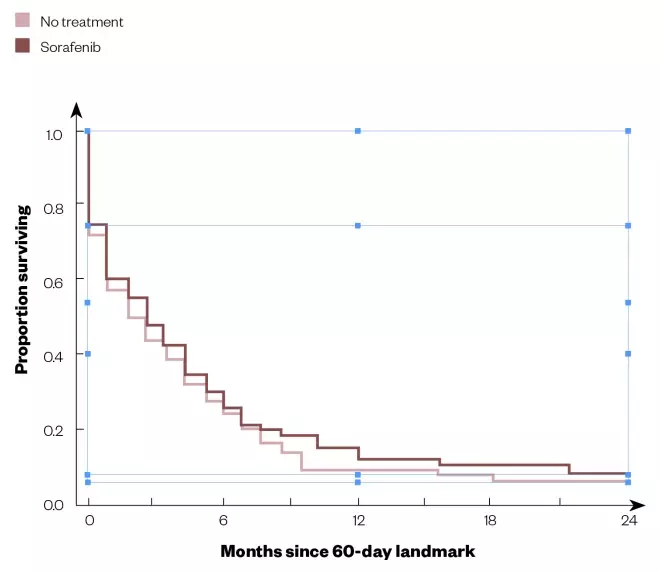
Figure 1: Survival curves for sorafenib treatment versus no treatment in real-world patients with advanced hepatocellular carcinoma
Source: Reproduced with permission from The Oncologist
[22]
Kaplan–Meier survival estimates from 60-day landmark of propensity score-matched patients.
Pharmacokinetic barriers related to oral bioavailability
A major problem with many anticancer drugs is their low and variable oral bioavailability. Average absolute bioavailabilities (F) of a range of compounds are shown in Figure 2. While there are some compounds that exhibit complete bioavailability, the range extends well down to virtually zero. In many cases, estimates of oral bioavailability of compounds that are used in this way are not available, reflecting a reluctance to carry out intravenous studies on new compounds intended for oral use only, even though this can be done safely using a micro-dose or nano-dose and allows an understanding of the reason (or reasons) for low bioavailability.

Figure 2: The mean oral bioavailabilities of some anticancer medicines
Source: Values taken from DrugBank
‘F’ indicates the fraction or percentage of the dose that reaches the systematic circulation as intact drug.
A low oral bioavailability might be circumvented simply by giving a larger dose. However, this may not be possible with some drugs, such as etoposide, if the absorption is saturable[1]
or there is limiting toxicity. The main problem with a low oral bioavailability is an increased variability in systemic exposure[10]
.
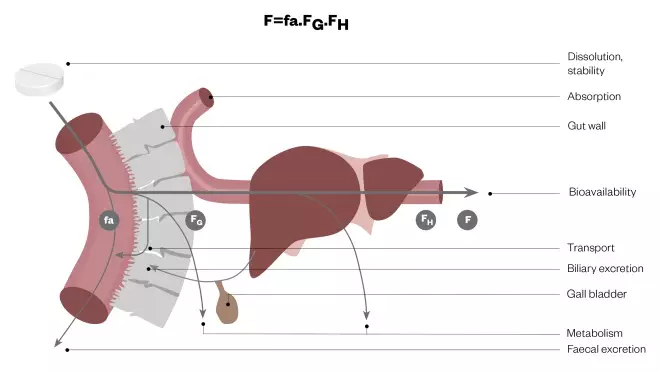
Figure 3: Elements of oral bioavailability
Net bioavailability (F) is seen to be the running product of fa (the fraction of the dose released from the dosage form that dissolves in the gastrointestinal fluid and enters the enterocyte), FG (the fraction that avoids first-pass metabolism and/or efflux transport in the gut wall and is transferred to the liver) and FH (the fraction that avoids first-pass metabolism in the liver and gains entry to the systematic circulation).
Net oral bioavailability (Figure 3) is the running product of:
- fa — the fraction of the dose released from the dosage form that dissolves in the gastrointestinal fluid and enters the enterocyte;
- FG — the fraction that avoids first-pass metabolism in the gut wall and is transferred to the liver;
- FH — the fraction that avoids first-pass metabolism in the liver and gains entry to the systemic circulation.
A low fa will reflect sub-optimal physicochemical properties and the activity of efflux transporters, FG, will reflect the combined activity of enzymes and efflux transporters as biological barriers and FH will reflect the activity of enzymes and biliary transporters.
Physicochemical properties
Important considerations in the design of drugs for oral use include lipid solubility (as it affects passive permeability across the enterocyte), chemical stability in the gut lumen and aqueous solubility (as it affects dissolution and potential re-precipitation in the gut lumen). Accordingly, structural features contributing to a sub-optimal log P value[11]
(representing the partition of drug between octanol and aqueous buffer at a pH of 7.4 as a measure of lipid solubility), poor chemical stability and low aqueous solubility will predict a low fa value.
If the log P value is below around –0.4, the compound will have difficulty diffusing across the enterocyte. However, this will be facilitated if the log P value is above around 5, albeit at the expense of a greater difficulty in getting into the solution in the gut lumen. Many oral anticancer drugs have log P values either above or below this optimal range.
Poor chemical stability in gastric and intestinal fluids is a primary cause of the low and variable oral bioavailability of etoposide and chlorambucil, for example[7]
.
The biggest issue in oral bioavailability is often poor aqueous solubility. Although this tends to be inversely related to lipid solubility, medicinal chemists make a distinction between ‘brickdust’ and ‘grease’. While compounds in both categories exhibit low aqueous solubility, those in the former group have a lower lipid solubility than the latter, which might limit their ability to diffuse across the gut wall. The sTKIs have aqueous solubilities that are similar to limestone and quartz (see Figure 4), although they do have reasonable log P values. Thus, their low aqueous solubility rather than a problematic lipid solubility is likely to contribute to the incomplete and variable oral bioavailability of some of these compounds[8],[9]
.
The balance between membrane permeability and aqueous solubility is reflected in the Biopharmaceutics Classification Scheme[12]
, which defines four permutations:
- Class 1: high permeability and solubility;
- Class 2: high permeability and low solubility;
- Class 3: low permeability and high solubility;
- Class 4: low permeability and solubility.
Most anticancer drugs fall into classes 2, 3 or 4, with properties not conducive to good oral availability because of low solubility, low permeability or both.
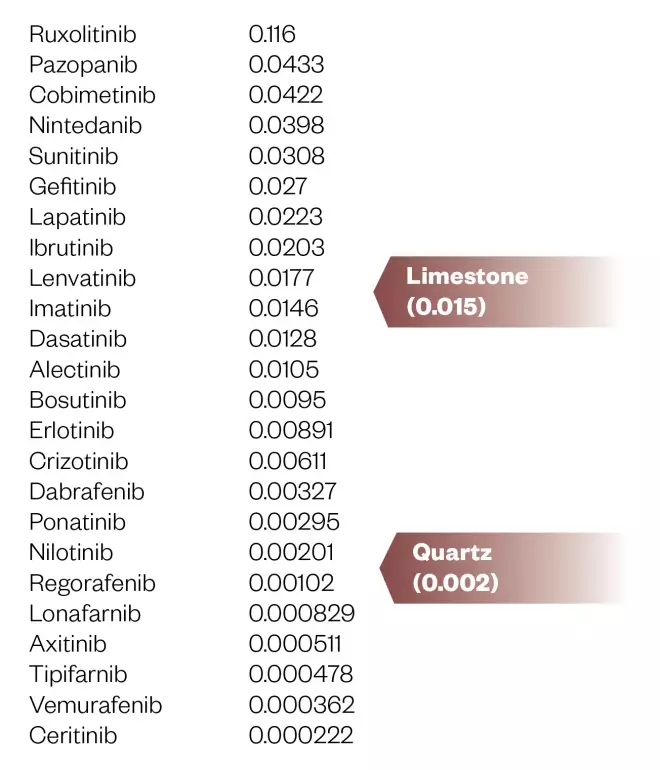
Figure 4: Aqueous solubility (mg/mL) of small molecule tyrosine kinase inhibitors (predicted or experimental)
Data taken from DrugBank
First-pass metabolism and efflux transport
Many anticancer drugs undergo significant first-pass metabolism in the gut wall, the liver or both. 5-fluorouracil (5FU) has an erratic oral bioavailability because of first-pass metabolism by dihydropyrimidine dehydrogenase (DPD) and sTKIs, with the exception of afatinib, which does not appear to be metabolised at all, are to varying extents substrates of intestinal and hepatic CYP3A[7],[8],[9],[13]
.
Compounding the impact of sub-optimal physicochemical properties and first-pass metabolism on the extent of oral availability is the fact that many anticancer drugs are also substrates of efflux transporters in the gut wall, particularly P-glycoprotein (ABCB1), the multidrug-resistance associated proteins MRP2 and 4 (ABCCCs) and the breast cancer resistance protein BCRP (ABCG2)[7],[8]
. Efflux back into the gut lumen will augment exposure to gut wall enzymes following any reabsorption. These transporters are also responsible for the efflux drugs from tumours and their up-regulation leads to resistance to therapy.
Potential solutions to low oral bioavailability
A number of strategies are used to address the problem of poor oral bioavailability, including the development of pro-drugs, the use of deliberate drug–drug interactions involving inhibition of enzymes or efflux transporters, and formulation in sophisticated carrier systems. While these approaches continue to be investigated, formal regulatory approval lags significantly behind innovation.
Pro-drugs
Pro-drugs can improve oral bioavailability by increasing the aqueous solubility or chemical stability of the active drug, or by protecting it from pre-systemic metabolism[7]
. The phosphate salt of etoposide and the mesylate of imatinib increase solubility and enhance oral bioavailability significantly. While MTIC (3-methyl-(triazen-1-yl) imidazole-4-carboxamide), a DNA alkylating agent, is highly unstable in gastric fluid, the pro-drug temozolomide is stable at low pH and breaks down rapidly to MTIC by a non-enzymatic chemical reaction at physiological pH. Doxifluridine, capecitabine and tegafur are precursors of 5FU, which are not cleavable by DPD but utilise other enzymes on first-pass through the liver to convert them into the active drug. Capecitabine has been shown to be as effective as the ‘Mayo Clinic IV regimen’ for 5FU[1]
.
Co-administration with inhibitors of metabolism or efflux transport
The addition of ritonavir (a potent inhibitor of CYP3A) to paclitaxel and docetaxel significantly increases their oral bioavailability while, similarly, the combination of gimeracil, uracil or eniluracil (inhibitors of DPD) with 5FU enhances its systemic exposure[7]
. However, eniluracil has proved to be disappointing in a clinical trial[1]
. Cyclosporine and elacridar are potent inhibitors of P-glycoprotein and their combination with paclitaxel or topotecan is accompanied by significant gain in the systemic exposure of these agents after oral administration[7]
.
While the production of a deliberate drug–drug interaction is effective in raising oral bioavailability, it carries the risk of creating unwanted interactions with other drug or nutrient substrates of the affected enzyme or transporter. By knocking out P-glycoprotein at the blood–brain barrier, co-administration of an inhibitor of this transporter could enhance access of the victim drug to a brain tumour. Although, equally, it may increase the likelihood of central nervous system side effects from the agents involved.
Formulation in carrier systems
Current research on carrier systems, especially those involving nanotechnology, is flourishing. The objective, with respect to enhancing oral bioavailability, is to improve absorption while getting ‘under the radar’ of exposure to first-pass enzymes and efflux transporters. In doing this, a number of absorption mechanisms as alternatives to transcellular passive diffusion are exploited, including the opening of tight intercellular junctions, endocytosis and pinocytosis, lipid absorption and M-cell uptake[7],[14]
(see Figure 5). Notably, some of these mechanism’s direct delivery via the lymphatic system rather than straight into the blood circulation, circumventing significant first-pass metabolism.
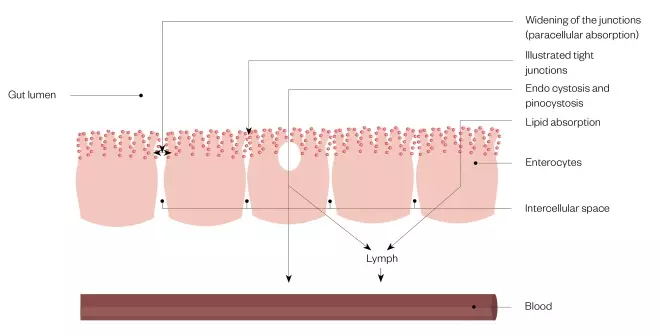
Figure 5: Absorption mechanisms exploited by drug formulations designed to enhance bioavailability
As an alternative to transcellular diffusion across the enterocyte into the blood, access to systemic exposure may be gained by widening of the tight intercellular junctions between the enterocytes to allow enhanced paracellular absorption, endo- or pino-cytosis or by exploitation of absorption pathways for lipids. The latter routes deliver drug into the lymph.
Many different types of carrier formulations are being explored (see Figure 6) and have been reviewed comprehensively elsewhere[7],[14]
. It is worth noting that while lipid-based formulations can enhance the absorption of ‘greasy’ compounds, they may not do so for ‘brickdust’[15]
. Carrier formulations include smart systems that focus delivery of their drug payload at the tumour target, rather than only into the systemic circulation or tissues in general. Time will tell if they will reach commercialisation since progress in this respect is challenged by issues of manufacturing scale-up and product stability[16],[17]
(especially under home storage conditions and on the way to target) and the ultimate barrier of regulatory approval (especially with regard to the toxicology and safety of the carrier materials used[18],[19]
).
The really big challenge is to deliver biologics orally through nanotechnology[20]
. Although advances have been made in delivering insulin in this way, the drive to do this for monoclonal antibodies is perhaps less because they have inherently long durations of effect, requiring only infrequent dosing.
It also remains to be seen whether bioengineering advances in the development of wearable injector devices[21]
will take precedence over pharmaceutical formulations, as practical solutions to improving the home delivery of anticancer drug treatment.
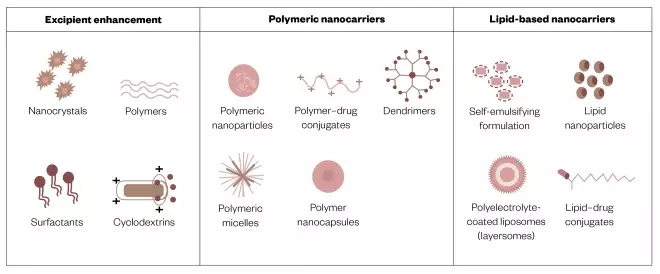
Figure 6: Formulation strategies for the oral delivery of anticancer drugs
Source: Modified with permission from J Contr Rel
[14]
Conclusion
Oral anticancer drug treatment is often compromised by low and variable bioavailability as a consequence of deficient physicochemical properties and first-pass metabolism and efflux. Research and development to overcome these difficulties is a major element of contemporary pharmaceutical science, with the aim of improving the prospect of cancer chemotherapy in the home.
Financial and conflicts of interest disclosure
The author has no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. No writing assistance was used in the preparation of this manuscript.
Acknowledgements
This article is based on a presentation at the 78th International Pharmaceutical Federation World Congress, Glasgow, 2–6 September 2018.